


ЭЛЕКТРОГРАВИМЕТРИЯ, электрохим.
метод количеств, анализа, основанный на определении увеличения массы рабочего
электрода вследствие выделения на нем определяемого компонента в результате
электролиза. Как правило, определяемое в-во осаждают в виде металла (или
оксида) на предварительно взвешенном платиновом катоде (или аноде). Момент
завершения электролиза устанавливают с помощью специфич. чувствительной
качественной р-ции на определяемый ион. Рабочий электрод промывают, высушивают
и взвешивают. По разности масс электрода до и после электролиза определяют
массу выделившегося металла или оксида.
Теоретич. потенциал выделения металла
на катоде можно рассчитать из величин стандартных электродных потенциалов
Е0.
Напр., при определении Cu(II) в кислом р-ре на платиновых катоде и аноде
протекают соотв. р-ции:
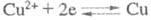 (
(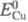 =0,339 В)
и
=0,339 В)
и
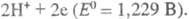 В условиях электролиза потенциал катода при 25 °С описывается ур-нием Нернста:
В условиях электролиза потенциал катода при 25 °С описывается ур-нием Нернста:
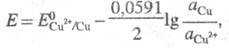
где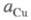 и
и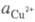 - термодинамич.
активности соотв. металлической меди и ионов
- термодинамич.
активности соотв. металлической меди и ионов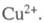 В начале электролиза, когда пов-сть катода не покрыта медью,
В начале электролиза, когда пов-сть катода не покрыта медью,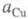 бесконечно малая величина; при наличии тока, достаточного для заполнения
медью пов-сти катода,
бесконечно малая величина; при наличии тока, достаточного для заполнения
медью пов-сти катода,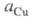 приближается
к единице. На практике для протекания электрохим. р-ции с заметной скоростью
необходимо более высокое напряжение, чем теоретически рассчитанный потенциал
выделения Е. Это связано с перенапряжением кислорода на платиновом
аноде (порядка 0,84 В при плотности тока 0,01 А х см-2) и омич.
падением напряжения в ячейке.
приближается
к единице. На практике для протекания электрохим. р-ции с заметной скоростью
необходимо более высокое напряжение, чем теоретически рассчитанный потенциал
выделения Е. Это связано с перенапряжением кислорода на платиновом
аноде (порядка 0,84 В при плотности тока 0,01 А х см-2) и омич.
падением напряжения в ячейке.
Э.- селективный метод: при равенстве исходных
концентраций компонентов раздельное выделение на электроде возможно при
разности их электродных потенциалов порядка 0,3 В (для однозарядных ионов)
или 0,1 В (для двухзарядных ионов).
Электролиз можно проводить при постоянном
напряжении между электродами, при постоянной силе тока или при контролируемом
потенциале рабочего электрода. В случае Э. при постоянном напряжении происходит
смещение потенциала рабочего электрода в более отрицат. область за счет
поляризации. Следствием этого является снижение селективности из-за протекания
дополнит. р-ции (выделение др. металлов или газообразного Н2).
Этот вариант Э. пригоден для определения легко восстанавливающихся в-в
в присут. примесей, восстанавливающихся труднее, чем ионы Н+.
В конце электролиза возможно выделение газообразного Н2. Хотя
в отличие рт кулонометрии 100%-ный выход по току определяемого в-ва не
обязателен, выделение Н2 часто приводит к образованию осадков
с неудовлетворительными физ. св-вами. Поэтому в анализируемый р-р рекомендуется
вводить в-ва, восстанавливающиеся легче ионов Н+ (гидразин,
гидроксиламин) и предотвращающие т. обр. выделение Н2.
Если проводить электролиз при постоянной
силе тока, необходимо периодически увеличивать налагаемое на ячейку внешнее
напряжение, чтобы скомпенсировать уменьшение тока, вызываемое концентрационной
поляризацией. Вследствие этого анализ становится менее селективным. Иногда,
однако, удается связывать мешающие катионы в прочные комплексные соед.,
восстанавливающиеся при более отрицат. потенциале, чем определяемое в-во,
или предварительно удалять мешающий ион в виде малорастворимого соединения.
Метод применяют, напр., для определения Cd в щелочном р-ре его цианида,
Со и Ni в аммиачно-сульфатном р-ре, Си в смеси серной и азотной к-т.
Значительно более высокая селективность
достигается в случае проведения электролиза при контролируемом потенциале
рабочего электрода. Обычно потенциал рабочего электрода измеряют относительно
третьего электрода с известным и постоянным потенциалом, т. е. электрода
сравнения (насыщенного каломельного или хлорсеребряного). Имеются потенциостаты,
поддерживающие постоянный потенциал катода на протяжении всего электролиза.
При этом можно раздельно количественно выделять компоненты смеси со стандартными
электродными потенциалами, различающимися всего на неск. десятых долей
вольта. Напр., последовательно определяют Сu, Bi, Pb и Sn. Первые три металла
выделяют из нейтрального тартратного р-ра: Сu - при 0,2 В; Bi - при 0,4
В; Рb - при 0,6 В (взвешивая электрод после осаждения каждого металла).
Оставшийся р-р подкисляют и осаждают Sn при -0,65 В. Разработаны методы
определения Сu в присут. Bi, Sb, Pb, Sn, Ni, Cd, Zn; Pb - в присут. Cd,
Sn, Ni, Zn, Mn, Al, Fe.
Выделенный на электроде осадок должен
хорошо прилипать к электроду, быть плотным и гладким во избежании мех.
потерь при промывании, высушивании и взвешивании. На физ. св-ва осадков
влияют плотность тока, т-ра и интенсив-' ность перемешивания р-ра. Электролиз
рекомендуется проводить при невысоких плотностях тока (обычно от 0,01 до
0,1 А x см-2), что позволяет получать мелкокристаллич., свободные
от примесей осадки, хорошо удерживающиеся на электроде, а также избегать
концентрационной поляризации. Для снижения плотности тока применяют рабочие
электроды с большой пов-стью (в частности, сетчатые); одновременно в этом
случае сокращается и время анализа. Для снижения концентрационной поляризации
и ускорения электролиза р-р интенсивно перемешивают или, иногда, нагревают.
В последнем случае увеличивается подвижность ионов и уменьшается вязкость
р-рителя; но одновременно может усилиться выделение газообразных продуктов,
поэтому для каждого конкретного определения оптимальную т-ру устанавливают
экспериментально.
Разновидность Э.- метод внутр. (самопроизвольного)
электролиза, когда электрохим. р-ция в ячейке (гальванич. элементе) протекает
самопроизвольно без приложения внеш. напряжения. Катодом служит инертный
металлич. электрод (обычно платиновая сетка), анодом - электрохимически
активный электрод, напр. пластинка из меди, цинка или магния. Электролиз
начинается в момент соединения электродов внеш. проводником и проходит
до тех пор, пока полностью не выделится определяемый металл. Для поддержания
относительно высокой силы тока применяют электроды большого размера, хорошо
перемешивают р-р, вводят инертный электролит. Чтобы избежать выделения
определяемого в-ва на аноде (цементация), анодное пространство отделяют
от катодного пористой диафрагмой или анод изолируют от анализируемого р-ра
с помощью пористого керамич. стаканчика, заполненного р-ром соли металла,
из к-рого изготовлен анод. При правильном выборе анода можно проводить
селективные определения. Напр., с платиновым катодом и медным анодом в
р-ре сульфата меди определяют Ag в присут. Си, Fe, Ni и Zn. В общем случае
при катодном выделении определяемого в-ва потенциал анода должен быть отрицательнее
потенциала рабочего электрода. Метод внутр. электролиза более пригоден
для определения сравнительно малых кол-в в-ва, отличается простотой и селективностью;
недостаток метода - длительность анализа (для полного выделения осадка
необходимо вести электролиз не менее часа).
Э. известна с 1860-х гг. и применялась
для определения металлов, используемых для чеканки монет, в разл. сплавах
и рудах. Это безэталонный метод, к-рый можно рассматривать как простейший
вариант кулонометрии. По точности и воспроизводимости результатов Э. превосходит
др. методы при определении таких металлов, как Сu, Sn, Pb, Cd, Zn. Несмотря
на относит, длительность эксперимента, Э. до сих пор применяют для анализа
сплавов, металлов и р-ров для электролитных ванн.
Лит.: Лопатин Б. А., Теоретические основы электрохимических методов анализа, М., 1975; Скуг Д., Уэст Д., Основы аналитической химии, пер. с англ., т. 2, М., 1979; Юинг Г., Инструментальные методы химического анализа, пер. с англ., М., 1989.
Г. В. Прохорова.