


Неподвижная фаза гл. обр. представляет собой сорбент с развитой пов-стью, а подвижная - поток газа (пара, флюида -в-во в сверхкритич. состоянии) или жидкости. Поток подвижной фазы фильтруется через слой сорбента или перемещается вдоль слоя сорбента.
Основные виды хроматографии.
В зависимости
от агрегатного состояния подвижной фазы различают газовую, флюидную (или
сверхкритич. X. с флюидом в качестве элюента; см. Капиллярная хроматография)и жидкостную X. В качестве неподвижной фазы используют твердые (или
твердообразные) тела и жидкости. В соответствии с агрегатным состоянием
подвижной и неподвижной фаз различают следующие виды X.: 1) газо-твердофазную
X., или газоадсорбционную хроматографию; 2) газо-жидкостную хроматографию
(газо-жидко-твердофазную); 3) жндко-твердофазную X.; 4) жидко-жидкофазную
X.; 5) флюидно-твердофазную X.; 6) флюидно-жидко-твердофазную X.
Строго говоря, газо-жидкостная X. пока
не реализована, на практике используют только газо-жидко-твердо-фазную
X. (см. Газовая хроматография). Жидко-жидкофазная X. реализована,
однако преим. используют жидко-жидко-твердо-фазную X. (неподвижной фазой
служит твердый носитель с нанесенной на его пов-сть жидкостью; см. Жидкостная
хроматография).
По механизму разделения в-в различают
адсорбционную, распределительную, ионообменную, эксклюзионную, аффинную
(биоспецифическую), осадочную X. На практике часто реализуется одновременно
неск. механизмов разделения (напр., адсорбционно-распределителъный, адсорбционно-эксклюзионный
и т. д.).
По геометрии сорбционного слоя неподвижной
фазы различают колоночную и плоскослойную X. К плоскослойной относятся
тонкослойная хроматография и бумажная хроматография.
В колоночной
X. обычно выделяют капиллярную X., в к-рой сорбент расположен на внутр.
стенках колонки, а центр, часть колонки остается незаполненной сорбентом,
т.е. открытой для потока элюента (X. на открытых капиллярных колонках).
В зависимости от способа ввода пробы и
способа перемещения хроматографич. зон по слою сорбента различают след.
варианты X.: проявительный (или элюентный), фронтальный и вытеснительный.
В наиб. часто используемом проявительном варианте анализируемую смесь периодически
импульсно вводят в поток подвижной фазы; в колонке анализируемая смесь
разделяется на отдельные компоненты, между к-рыми находятся зоны подвижной
фазы.
Во фронтальном варианте X. пробу, содержащую
разделяемые в-ва, непрерывно подают в колонку. Можно также подавать в колонку
одновременно пробу и подвижную фазу. Во фронтальной X. только первый, наименее
сорбируемый компонент можно получить в чистом виде на выходе из колонки,
вторая и последующая зоны содержат по два и более компонентов разделяемой
смеси.
В вытеснительном варианте X. в колонку
после подачи разделяемой смеси вводят спец. в-во (т. наз. вытеснитель),
к-рое сорбируется лучше любого из разделяемых компонентов. В вытеснительной
X. образуются примыкающие друг к другу зоны разделяемых в-в. Во фронтальном
и вытеснительном вариантах X. необходима регенерация колонки перед след.
опытом.
Основы хроматографич. процесса. Для
проведения хроматографич. разделения в-в или определения их физ.-хим. характеристик
обычно используют спец. приборы - хроматографы. Осн. узлы хроматографа
- хроматографич. колонка, детектор, а также устройство для ввода пробы.
Колонка, содержащая сорбент, выполняет ф-цию разделения анализируемой смеси
на составные компоненты, а детектор -ф-цию их количеств. определения. Детектор,
расположенный на выходе из колонки, автоматически непрерывно определяет
концентрацию разделяемых соед. в потоке подвижной фазы (см. Детекторы
хроматографические).
После ввода анализируемой смеси с потоком
подвижной фазы в колонку зоны всех в-в расположены в начале хроматографич.
колонки (рис. 1). Под действием потока подвижной фазы компоненты смеси
начинают перемещаться вдоль колонки с разл. скоростями, величины к-рых
обратно пропорциональны коэффициентам распределения К (или константам
распределения) хроматографируемых компонентов. Хорошо сорбируемые в-ва,
значения констант распределения для к-рых велики, передвигаются вдоль слоя
сорбента по колонке медленнее, чем плохо сорбируемые. Поэтому быстрее всех
из колонки выходит компонент А, затем компонент Б и последним покидает
колонку компонент В (КА<КБ<КВ).
Сигнал
детектора, величина к-рого пропорциональна концентрации определяемого в-ва
в потоке элюента, автоматически непрерывно записывается и регистрируется
(напр., на диаграммной ленте). Полученная хроматограмма отражает расположение
хроматографич. зон на слое сорбента или в потоке подвижной фазы во времени.
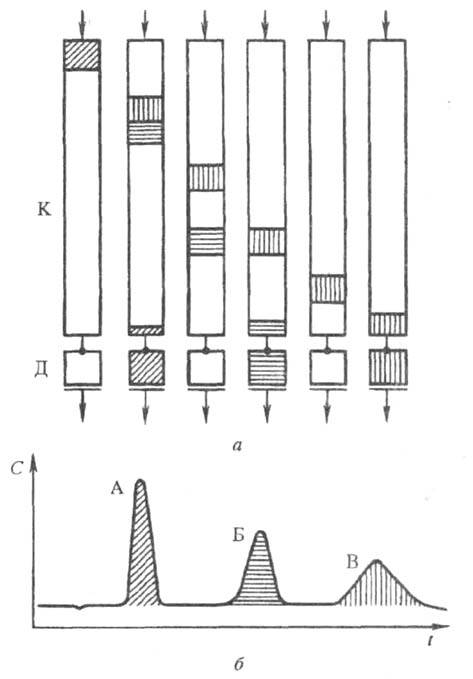
Рис. 1. Разделение смеси из трех компонентов (А, Б и В) на хроматографической колонке К с детектором Д: а - положение хроматографических зон разделяемых компонентов в колонке через определенные интервалы времени; б - хроматограмма (С - сигнал, t - время).
При плоскослойном хроматографич. разделении лист бумаги или пластину со слоем сорбента с нанесенными пробами исследуемого в-ва помещают в хроматографич. камеру. После разделения компоненты определяют любым подходящим методом.
Основные величины удерживания и качественный
анализ. Хроматограмма - первичный результат хроматографич. разделения.
Используя хроматограмму, можно определять осн. характеристики хроматографич.
процесса: параметры удерживания, размывания и разделения хроматографируемых
соединений. Осн. характеристика в-ва при колоночной X. (если т-ра колонки,
состав подвижной фазы и ее скорость постоянны) - объем удерживания (или
время удерживания в случае жидкостной А.), к-рый для i-го компонента
зависит от его константы распределения Ki.
Если неподвижная фаза - твердое тело,
на пов-сть к-рого нанесена в форме тонкого слоя неподвижная жидкая фаза
(НЖФ), удерживание определяется как абсорбцией разделяемых соед. слоем
НЖФ, так и их адсорбцией пов-стями раздела: подвижная фаза - НЖФ и НЖФ
- твердое тело. Для качеств. характеристики хроматографируемых соед. преим.
применяют относит. величины удерживания, поскольку эти величины в меньшей
мере, чем абс. величины, зависят от условий эксперимента.
Для характеристики относит. времени удерживания
в X. используют системы с двумя стандартами, в качестве к-рых в наиб. распространенной
системе индексов удерживания Ковача Ii применяют соед.
одного гомологич. ряда. Эти стандарты выбирают таким образом, чтобы определяемое
соед. выходило из колонки позже стандарта (напр., алкана), молекула к-рого
содержит z атомов углерода, и раньше стандарта, молекула к-рого
содержит z + 1 атомов углерода. Ii определяют по ф-ле
(рис. 2):
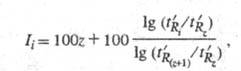
где -
время удерживания алкана
-
время удерживания алкана исправленные
времена удерживания соотв. для алканов Сz и Сz+1
и i-го компонента;
tm - время удерживания несорбирующегося
компонента.
исправленные
времена удерживания соотв. для алканов Сz и Сz+1
и i-го компонента;
tm - время удерживания несорбирующегося
компонента.
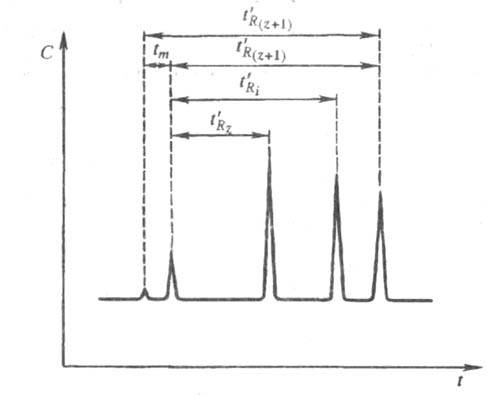
Рис. 2. Определение индекса удерживания Ii с использованием н-алканов с числом атомов z и z + 1; пояснения в тексте.
Идентификацию пиков неизвестных компонентов анализируемой смеси проводят путем сопоставления (сравнения) относит. величин, определяемых непосредственно из хроматограммы, с соответствующими табличными данными для известных соединений. При идентификации в X. достоверен только отрицат. ответ; напр., пик i не является в-вом А, если времена удерживания пика i и в-ва А не совпадают. Совпадение времен удерживания пика i и в-ва А - необходимое, но недостаточное условие для заключения, что пик i - это в-во А.
Эффективность хроматографической колонки.
При продвижении зон разделяемых соед. под действием потока подвижной
фазы вдоль слоя сорбента происходит одновременно два противоположных процесса:
возрастает расстояние между максимумами концентрации хроматографич. зон
(что улучшает разделение) и увеличивается ширина хроматографич. зон (что
ухудшает разделение). Качественно эффективность колонки тем выше, чем уже,
острее зоны хроматографируемых соединений. Количеств. характеристикой эффективности
колонки служит число теоретич. тарелок. Эффективность колонки тем выше,
чем больше характерное для нее число теоретич. тарелок N. Число
N для i-го компонента вычисляют по ур-нию: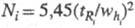 , где
, где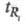 и
и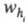 -
соотв. время удерживания i-го компонента и ширина пика, измеренная
на половине его высоты (рис. 3). Число N пропорционально квадрату
числа пиков, к-рые можно разместить на хроматограмме на отрезке, соответствующем
времени удерживания данного соединения.
-
соотв. время удерживания i-го компонента и ширина пика, измеренная
на половине его высоты (рис. 3). Число N пропорционально квадрату
числа пиков, к-рые можно разместить на хроматограмме на отрезке, соответствующем
времени удерживания данного соединения.
Разделение. Разделение смеси соед. - основная цель аналит. и препаративной X. Для характеристики разделения трудноразделимых (критических) пар соед. используют особую величину - степень разделения (рис. 3):
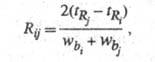
где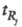 -
время удерживания j-гo компонента; - исправленное время удерживания
j-гo
компонента;
-
время удерживания j-гo компонента; - исправленное время удерживания
j-гo
компонента;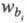
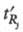 и
и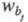 - ширина
хроматографич. зон, измеренная у основания пиков на хроматограмме,
- ширина
хроматографич. зон, измеренная у основания пиков на хроматограмме,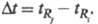
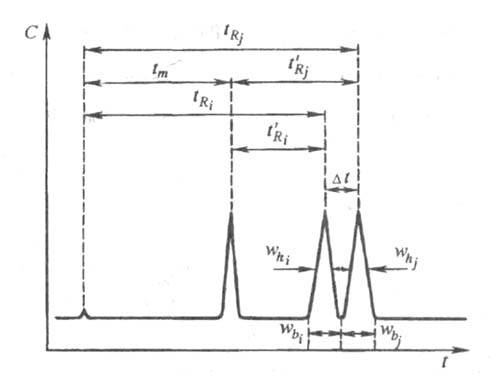
Рис. 3. Определение степени разделения Rij; пояснения в тексте.
Количественно зависимость степени разделения от параметров хроматографич. разделения отражает ур-ние Пернелла:
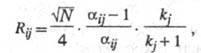
где aij
- селективность разделения i-го и j-гo компонентов;
kj
- коэф. емкости (или коэф. извлечения) компонента
j, причем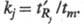 Как следует из этого ур-ния, степень разделения увеличивается с ростом
эффективности колонки
Как следует из этого ур-ния, степень разделения увеличивается с ростом
эффективности колонки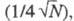 селективности (aij
-
1) и емкости колонки kj/(kj + 1). Селективность
разделения характеризуется величиной относит. удерживания
rij:
селективности (aij
-
1) и емкости колонки kj/(kj + 1). Селективность
разделения характеризуется величиной относит. удерживания
rij:
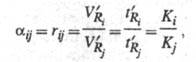
где VRj и VRi
- исправленный объем удерживания для j-гoи i-го компонентов;
Ki и Kj - коэф. распределения в системе
неподвижная фаза - подвижная фаза для
i-го и j-гo компонента.
Величины rij достаточно инвариантны; они не зависят от
таких условий эксперимента, как скорость газа-носителя, кол-во сорбента,
длина колонки и т. п.
X. - один из основных методов количеств.
анализа орг. и неорг. соединений. При постоянных условиях эксперимента
величина сигнала детектора прямо пропорциональна концентрации i-го
компонента в подвижной фазе, а площадь его пика на хроматограмме Si
- кол-ву анализируемого соединения. Долю i-го компонента в процентах
в n-компонентной смеси рассчитывают по ф-ле:
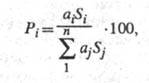
где аi и aj
- поправочные коэф., определяемые чувствительностью детектора к анализируемым
в-вам. Предел обнаружения при использовании высокочувствит. детекторов
составляет 10-10%, обычно погрешность определения 0,1-20%.
Недостаток хроматографич. методов - периодичность
анализа (показания запаздывают на время, равное продолжительности разделения)
- является существенным, в осн., для пром. X., к-рую используют для контроля
и регулирования пром. многотоннажных процессов.
Аналит. X. применяют в научных исследованиях,
хим. и фармацевтич. пром-сти, медицине, для контроля практически всех объектов
окружающей среды, в газовой и нефтеперерабатывающей пром-сти и т. д.
Препаративную X. используют для получения
узких фракций смесей и чистых в-в в произ-ве хим. реактивов, а также в
фармацевтич., парфюмерной пром-сти, при разделении изотопов, в биохимии
и т. п. Разделяют смеси массой 1-1000 г, диаметр колонок 2-100 см.
X. применяют для определения физ.-хим.
характеристик в-в: коэф. распределения, энтальпии растворения, адсорбции,
констант устойчивости комплексных соед., коэф. диффузии в газовой и жидкой
фазах и т. д., а также как метод исследования кинетики гетерогенных и гомогенных
р-ций. См. также Хроматография с программированием температуры, Хромато-масс-спектрометрия.
X. открыл М. С. Цвет в 1905.
Лит.: Беленький Б. Г., Виленчик Л. З., Хроматография полимеров, М., 1978; Киселев А. В., Яшин Я. И. .Адсорбционная газовая и жидкостная хроматография, М., 1979; Кирхнер Ю., Тонкослойная хроматографах, пер. с англ., т. 1-2, М., 1981; Березкин В. Г., Газо-жидко-твердофазная хроматография, М., 1986; Хроматография, под ред. Э. Хефгмана, пер. с англ., ч. 1-2, М., 1986; Даванков В. А., Навратил Дж., Уолтон X., Лигандообменная хроматография, пер. с англ., М., 1989; Го льберт К. А., Вигдергауз М. С, Введение в газовую хроматографию, 3 изд., М., 1990; Количественный анализ хроматографическим методом, под ред. Э. Кэца, пер. с англ., М., 1990; Препаративная жидкостная хроматография, под ред. Б. Бидлиншейера, пер. с англ., М., 1990; Сверхкритическая флюидная хроматография, под ред. Р. Смита, пер. с англ., М., 1991; SnyderL., Kirkland J., Introduction to modem liquid chromatography, 2 ed., N. Y., 1979.
В. Г. Березкин.