


ФОТОХИМИЧЕСКИЕ РЕАКЦИИ,
хим. р-ции, протекающие под действием света. Поглощение фотона с длиной
волны ~ 100-1500 нм, чему соответствует энергия 0,8-12,4 эВ (80-1200 кДж/моль),
вызывает квантовый переход молекулы в-ва из основного электронного состояния
в одно из возбужденных состояний или фотоионизацию - отщепление электрона и
образование катион-радикала. Возбужденные состояния молекул имеют отличную от
основного состояния электронную структуру и, как правило, более высокую реакционную
способность. Молекулы вступают в хим. р-ции, первичные продукты к-рых (ионы,
радикалы, изомеры) чаще всего оказываются нестабильными. Конечные продукты Ф.
р. появляются в результате обычных термич. р-ций, к-рые протекают либо непосредственно
с участием первичных частиц, либо как ряд последовательных хим. превращений.
Как правило, для молекул
с четным числом электронов при фотовозбуждении первоначально образуется возбужденное
синглетное состояние (с мультиплетностъю, равной 1). Ф. р. обычно протекает
из нижнего возбужденного синглетного состояния или из триплетного состояния
(мультиплетность 3), к-рое получается из возбужденного синглетного состояния
путем интеркомбинационной конверсии (изменения спина одного из электронов).
С хим. р-циями возбужденных
молекул конкурируют фо-тофиз. процессы: испускание фотона (флуоресценция или
фосфоресценция), внутренняя и интеркомбинационная конверсия в нижележащие электронные
состояния (триплетное или основное). Вследствие этих процессов времена жизни
возбужденных синглетных состояний обычно не превышают 10-8-10-9с.
Триплетные состояния в жидких р-рах обычно "гибнут" в результате
безызлучат. перехода и дезактивации (тушения) примесями (напр., кислородом);
времена их жизни не превышают 10-5 с. В "жестких" средах
(замороженных р-рах, полимерных матрицах), где процессы дезактивации замедляются,
времена жизни триплетных состояний могут достигать десятка секунд.
Квантовый выход Ф
первичного продукта Ф. р., образующегося из к.-л. возбужденного состояния, равен
отношению скорости этой Ф. р. к сумме скоростей всех фотофиз. и фотохим. процессов
гибели этого возбужденною состояния. Поскольку такие процессы м. б. как мономолекулярными,
так и бимолекулярными, сумму их скоростей выражают через сумму констант скорости
ki,- мономолекулярных (для р-ций первого порядка) и псевдомономолекулярных
(для р-ций второго порядка) процессов, при условии, что для бимолекулярных р-ций
концентрация [X] реагента в осн. электронном состоянии гораздо больше концентрации
возбужденных молекул. Если 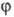 -
квантовый выход молекул в возбужденном состоянии (как правило,
-
квантовый выход молекул в возбужденном состоянии (как правило, 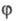 =
1 для возбужденных синглетных состояний и
=
1 для возбужденных синглетных состояний и 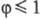 для триплетных состояний), кr - константа скорости рассматриваемой
Ф. р., то
для триплетных состояний), кr - константа скорости рассматриваемой
Ф. р., то
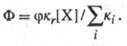
Суммарный выход конечного
продукта равен произведению выходов всех продуктов в ряду последоват. хим. превращений,
предшествующих образованию конечного продукта. В случае цепных
р-ций выход конечного продукта может значительно (иногда на неск. порядков)
превышать единицу.
Существует и др. определение
квантового выхода - как отношение числа молекул, участвующих в фотохим. или
фотофиз. процессе, к числу поглощенных фотонов. От квантового выхода следует
отличать квантовую эффективность -отношение скорости процесса к скорости образования
возбужденного состояния, из к-рого протекает данный процесс. Квантовый выход
и квантовая эффективность равны для процессов, происходящих из синглетного возбужденного
состояния.
Механизмы Ф. р. Большинство
Ф. р. протекает из терма-лизованных самых нижних возбужденных состояний соответствующей
мультиплетности (правило Каши). Это обусловлено чрезвычайно высокой скоростью
термализа-ц и и - установления термич. равновесия в результате перераспределения
избыточной колебат. энергии между разл. степенями свободы возбужденных молекул
и средой, а также высокой скоростью внутр. конверсии - переходов из высших возбужденных
состояний в низшие возбужденные состояния той же мультиплетности, к-рая значительно
превышает скорость большинства хим. р-ций и процессов испускания. Для таких
Ф. р. квантовый выход не зависит от энергии поглощаемого фотона (длины волны
возбуждающего света). Однако существуют также Ф. р., протекающие из нерелаксированных
(франк - кондоновских) возбужденных состояний, непосредственно образующихся
при поглощении фотона. Таковы, напр., нек-рые р-ции диссоциации и изомеризации.
При этом хим. р-ция конкурирует не с испусканием фотона или дезактивацией возбужденного
состояния, а с его релаксацией в состояние, из к-рого возможен переход с флуоресценцией
или фосфоресценцией. Квантовый выход таких р-ций не зависит от времени жизни
флуоресцентного или фосфоресцентного состояния, но зависит от энергии возбуждения.
Существует два принципиально
различных типа первичных р-ций фотовозбужденных молекул. При ад и а б а т и
ч. Ф. р. электронное возбуждение в элементарном хим. акте сохраняется, р-ция
полностью протекает на поверхности потенциальной энергии (ППЭ) возбужденного
электронного состояния и первичный продукт получается в возбужденном состоянии.
При диабатич. первичных р-циях (иногда неправильно наз. неадиабатическими) электронное
возбуждение в первичном хим. акте теряется, происходит переход с ППЭ возбужденного
состояния на ППЭ основного состояния и первичный продукт сразу же получается
невозбужденным. В нек-рых случаях и адиабатич. р-ции могут приводить к основному
состоянию продукта, если ППЭ основного и возбужденного состояний в области координаты
реакции, соответствующей первичному продукту, оказываются вырожденными (напр.,
при фотодиссоциации молекулы на атомы или радикалы).
Теория переходного состояния,
на к-рой основано большинство используемых в химии концепций реакционной способности
молекул в термич. р-циях (см. Активированного комплекса теория), применима
строго лишь к адиабатич. Ф. р. Диабатич. Ф. р. целесообразно рассматривать с
позиций теории безызлучат. переходов, однако она пока недостаточно развита,
особенно для сложных молекул. Наличие конкурирующих физ. процессов потери энергии
электронного возбуждения, константы скорости к-рых даже для изолир. молекул
очень велики и могут в ряде случаев достигать 1010 с-1
и более, осложняет механизм Ф. р. Способность фотовозбужденных молекул к
хим. взаимодействию определяется не столько значениями констант скорости соответствующих
хим. р-ций, сколько соотношением скоростей р-ции и конкурирующих с ней физ.
процессов деградации энергии электронного возбуждения. При взаимод. возбужденных
молекул с к.-л. реагентами, помимо процессов потери энергии возбуждения, присущих
самим возбужденным молекулам, возникают новые, часто еще более эффективные пути
деградации энергии, обусловленные появлением дополнит. степеней свободы молекулы
в реакционном комплексе и получившие назв. индуцированной дезактивации.
Для рассмотрения хим. активности
возбужденных молекул привлекают как "статич." представления об электронной
структуре (классификация мол. орбиталей, распределение электронной плотности,
эффективные заряды на атомах и т.п.), так и "динамич." характеристики,
т.е. изменения в элементарном хим. акте тех или иных параметров (сохранение
орбитальной симметрии, мультиплетности, изменение энергии Гиббса, энергии локализации
и др.).
Для Ф. р. наиб. перспективны
динамич. подходы, поскольку они позволяют учитывать специфику конкретных процессов
и в ряде случаев совместно рассматривать хим. превращение и конкурирующие с
ним процессы деградации энергии возбуждения. Р-ции возбужденных молекул с этой
точки зрения разделяют на разрешенные и запрещенные (по мультиплетности, орбитальной
симметрии и др.). Напр., при нарушении орбитальной симметрии на пути р-ции возникает
значит, потенциальный барьер, высота к-рого непосредственно не связана с энергетикой
р-ции. Скорость таких Ф.р. может сильно изменяться даже при слабых изменениях
структуры и симметрии молекул реагентов. Аналогично, для Ф. р., связанных с
изменением мультиплетности реагирующих частиц, весьма существенны факторы, влияющие
на спиновые взаимод. (см. Спин-орбитальное взаимодействие, Спин-спиновое
взаимодействие); эти факторы определяют вероятность интеркомбинационной
конверсии, к ним относится, в частности, наличие в реагирующих молекулах или
в среде тяжелых атомов парамагн. частиц, а также внеш. мага. поля.
Для адиабатич. Ф. р. и
диабатич. Ф. р., разрешенных правилами отбора (напр., для фотопереноса протона,
фотопереноса электрона, отрыва атома H и др.), наблюдаются однотипные зависимости
констант скорости от изменения энергии Гиббса в первичном фотохим. акте. Напр.,
для переноса электрона между возбужденной молекулой и донором электрона зависимость
энергии Гиббса активации 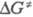 от энергии Гиббса переноса электрона
от энергии Гиббса переноса электрона 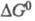 описывается ур-нием:
описывается ур-нием:
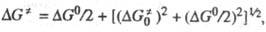
где 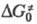 - эмпирич. параметр, соответствующий энергии активации изоэнергетич. р-ции (~
20 кДж/моль). Для нахождения
- эмпирич. параметр, соответствующий энергии активации изоэнергетич. р-ции (~
20 кДж/моль). Для нахождения 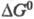 р-ций возбужденных молекул используют цикл Фёрстера, согласно к-рому в случае
адиабатич. р-ций энтальпия р-ции
р-ций возбужденных молекул используют цикл Фёрстера, согласно к-рому в случае
адиабатич. р-ций энтальпия р-ции 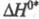 в возбужденном состоянии меньше энтальпии р-ции
в возбужденном состоянии меньше энтальпии р-ции  в основном состоянии на величину разности энергий возбуждения исходной молекулы
E* и первичного продукта
в основном состоянии на величину разности энергий возбуждения исходной молекулы
E* и первичного продукта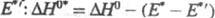 . Значения E* и E* ' легко определить из
эксперим. спектральных данных или расчетом. Для диабатич. Ф.р., где первичный
продукт образуется непосредственно в основном состоянии,
. Значения E* и E* ' легко определить из
эксперим. спектральных данных или расчетом. Для диабатич. Ф.р., где первичный
продукт образуется непосредственно в основном состоянии, 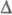 E*
' опускают. В большинстве случаев полагают, что энтропии р-ций в основном
и возбужденном состояниях примерно одинаковы и записывают аналогичное ур-ние
для
E*
' опускают. В большинстве случаев полагают, что энтропии р-ций в основном
и возбужденном состояниях примерно одинаковы и записывают аналогичное ур-ние
для  G0*,
к-рое позволяет вычислять константы равновесия и энергетику Ф. р. из возбужденных
состояний.
G0*,
к-рое позволяет вычислять константы равновесия и энергетику Ф. р. из возбужденных
состояний.
При классификации Ф. р.,
помимо общепринятых признаков - по типам разрываемых и образуемых связей, важное
значение имеют механизмы разрыва и образования связей.
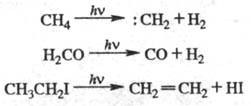
Основные типы Ф.р. Фотодиссоциация-
распад молекулы по к.-л. связи на радикалы, атомы или ионы, напр.:
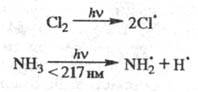
Фотодиссоциация происходит
при отталкивательном (диссо-циативном) типе ППЭ возбужденных состояний молекул.
В оптич. спектрах поглощения для переходов в "диссоциатив-ные" состояния
характерны сплошные полосы. Фотодиссоциация типична для таких возбужденных состояний,
в к-рых электрон находится на разрыхляющей s*-орбитали. Существуют
р-ции, когда первичный продукт образуется
в возбужденном состоянии [напр.,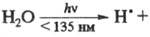
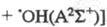 . В конденсир. средах первичные продукты фотодиссоциации оказываются в "клетке",
образованной частицами р-рителя, и могут рекомбинировать с образованием исходных
молекул (см. Клетки эффект), что приводит к существенному снижению квантового
выхода по сравнению с Ф. р. в газовой фазе, где выход часто близок к единице.
Фотодиссоциация - первичная стадия мн. р-ций замещения, стадия инициирования
в цепных р-циях.
. В конденсир. средах первичные продукты фотодиссоциации оказываются в "клетке",
образованной частицами р-рителя, и могут рекомбинировать с образованием исходных
молекул (см. Клетки эффект), что приводит к существенному снижению квантового
выхода по сравнению с Ф. р. в газовой фазе, где выход часто близок к единице.
Фотодиссоциация - первичная стадия мн. р-ций замещения, стадия инициирования
в цепных р-циях.
Предиссоциация - разновидность
фотодиссоциации, при к-рой после поглощения фотона молекула первоначально оказывается
в стабильном возбужденном состоянии, а из него переходит в диссоциативное возбужденное
состояние, напр.:
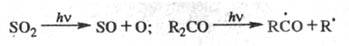
Фотодиссоциация может протекать
гомолитически либо гетеролитически. Из р-ций гетеролитич. фотодиссоциации наиб.
важны фотопротолитич. р-ции (связанные с гетеролитич. разрывом связи элемент
- водород). Известны также многочисленные р-ции гетеролитич. фотодиссоциации
по др. связям, в частности С — С, напр, в лейкооснованиях - триарилметановых
и нек-рых др. красителях: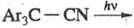
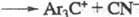 . Нек-рые из таких Ф. р. могут протекать адиабатически с образованием возбужденных
ионов красителей. Первоначально образующиеся карбкатионы могут взаи-мод. с нуклеофилами,
приводя в конечном итоге к продуктам нуклеоф. замещения. Широко распространена
гетеролитич. диссоциация в координационных соед., также приводящая в конце концов
к замещению лигандов.
. Нек-рые из таких Ф. р. могут протекать адиабатически с образованием возбужденных
ионов красителей. Первоначально образующиеся карбкатионы могут взаи-мод. с нуклеофилами,
приводя в конечном итоге к продуктам нуклеоф. замещения. Широко распространена
гетеролитич. диссоциация в координационных соед., также приводящая в конце концов
к замещению лигандов.
Распад (отщепление, фрагментация)-
разложение на мол. фрагменты, сопровождающиеся перегруппировкой связей, напр.:
Разрыв и перегруппировка
связей обычно происходят синхронно при движении системы частиц по соответствующей
сложной формы ППЭ возбужденного состояния. Такие р-ции подчиняются правилам
отбора для согласованных р-ции (см. Вудворда - Хофмана правила). Правило
сохранения орбитальной симметрии разрешает для возбужденных состояний, в отличие
от основного состояния, р-ции, протекающие через четырехцентровые переходные
состояния. Для таких р-ций характерно слабое влияние конденсир. фазы и присутствия
добавок, в частности акцепторов радикалов, на квантовый выход.
Фотоизомеризация. В зависимости
от характера изомеризации различают стереоизомеризацию, таутомерные превращения,
перегруппировки. Широко распространены процессы цис-транс- и транс-цис-фотоизомеризации
непредельных соед., напр. арилэтиленов и тиоиндиго:
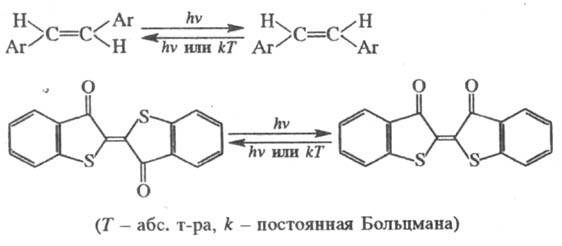
Эти Ф. р. обусловлены тем,
что минимумы на ППЭ возбужденных состояний, в отличие от ППЭ основного состояния,
часто соответствуют ортогональной, а не планарной конфигурации молекулы.
Практич. применение в качестве
фотохромных систем находит фотоизомеризация opmo-нитроароматич. соед.
и спи-ропиранов в мероцианины:
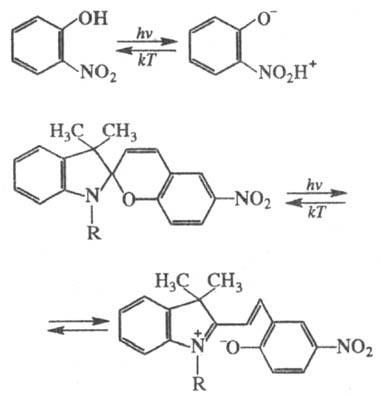
Широко распространены фотоперегруппировки
в ряду аро-матич. и циклич. непредельных соед., напр. бензола и его производных
- в бензвален, фульвен, т. наз. дьюаровский бензол и призман:
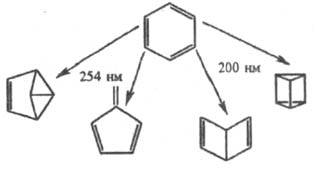
Окислительно-восстановит.
Ф.р. В основе большинства из них лежит фотоперенос электрона. Образующиеся в
первичной стадии ион-радикалы вступают в дальнейшие превращения, давая продукты
окисления или восстановления. Напр., при взаимод. дурохинона с донорами электрона
(аминами, спиртами) под действием света первоначально образуются семихиноновые
анион-радикалы, диспропорционирова-ние к-рых дает хинон и гидрохинон. Подобным
образом происходит фотовосстановление красителей (акридиновых, оксазиновых,
тиазиновых) до лейкоформ. Аналогично из ароматич. углеводородов RH в присут.
доноров электрона D получаются анион-радикалы, к-рые в протонных р-рителях присоединяют
протон и дают в конце концов продукты диспропорционирования, рекомбинации и
т. п.:
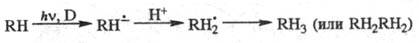
В координац. соед. часто
наблюдается фотоперенос электрона между центр. ионом и лигандом, что приводит
к образованию окисленной и восстановленной форм, напр.:
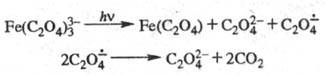
Перенос электрона может
происходить не только при взаимод. возбужденных молекул с донором или акцептором
электрона, но и путем прямой фотоионизации молекул. Для фотоионизации
требуется, чтобы энергия фотона превышала потенциал ионизации, что обычно существенно
больше, чем для возбуждения молекулы. В конденсир. фазе энергия, необходимая
для фотоионизации, понижается по сравнению с газовой фазой на 1-2 эВ вследствие
поляризации среды образующимися ионами. При фотоионизации (напр., аминов в замороженных
р-рах) оптич. и радиоспектроскопич. методами наблюдается образование их катион-радикалов.
Электрон первоначально
сольватируется р-рителем, а затем присоединяется к к.-л. акцептору, присутствующему
в р-ре.
Фотовосстановление и фотоокисление
могут протекать и не через стадии фотопереноса электрона. Так, фотовосстановление
карбонильных, гетероциклич. и нитроароматич. соед. происходит путем отрыва 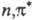 -возбужденными
состояниями этих молекул атома H от р-рителя и дальнейших превращений образующихся
радикалов, напр.:
-возбужденными
состояниями этих молекул атома H от р-рителя и дальнейших превращений образующихся
радикалов, напр.:
Фотоокисление кислородом
часто протекает путем фото-сенсибилизир. превращения мол. кислорода, основное
состояние к-рого является триплетным  , в синглетное
, в синглетное 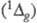 состояние. Синглетный кислород легко присоединяется по кратным связям и внедряется,
напр., по связи C-H:
состояние. Синглетный кислород легко присоединяется по кратным связям и внедряется,
напр., по связи C-H:
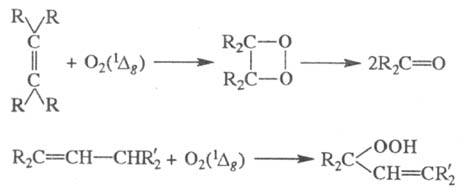
Присоединениек возбужденным
молекулам разл. реагентов характерно для многих ненасыщенных соед. Такие Ф.
р. обычно протекают по синхронному механизму и подчиняются соответствующим правилам
отбора (по мультиплет-ности, орбитальной симметрии и др.). Типичные примеры
-образование цйклобутановых соед. (а), оксетанов (б), фото-димеризация
(в), образование оксидов ароматич. соед. (г):
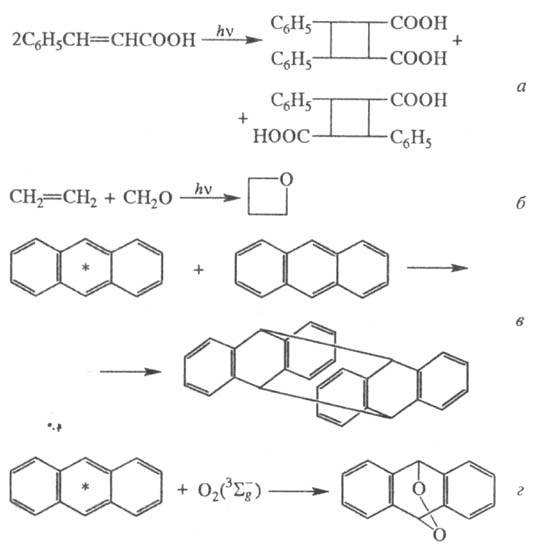
Известны процессы фотоприсоединения,
протекающие по радикальному (иногда цепному) механизму, напр.:
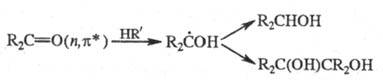
Отрыв атомов (гомолитический)
возбужденными молекулами от реагента (или р-рителя) характерен для возбужденных
состояний, имеющих неспаренный электрон на несвязывающей орбитали (напр., для
n, 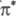 -состояний
карбонильных и гетероциклич. соед.):
-состояний
карбонильных и гетероциклич. соед.):
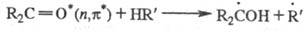
Первично образующиеся радикалы
вступают во вторичные р-ции рекомбинации или диспропорционирования, что приводит
к стабильным конечным продуктам (в данном примере пинаконам или спиртам - продуктам
восстановления исходного кетона). Причиной такого хим. поведения возбужденных
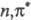 -состояний
является сходство их электронной структуры со структурой радикалов. Для радикальных
р-ций типичны линейные зависимости логарифма константы скорости отрыва от энергии
разрываемой связи.
-состояний
является сходство их электронной структуры со структурой радикалов. Для радикальных
р-ций типичны линейные зависимости логарифма константы скорости отрыва от энергии
разрываемой связи.
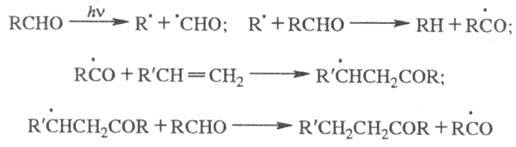
Внутримол. р-ции отрыва
атома водорода характерны для карбонильных соед. с достаточно длинными (более
двух атомов углерода) заместителями:

Промежут. бирадикал распадается
на два непредельных фрагмента - олефин и енол, последний затем изомеризуется
в кетон. Эта р-ция носит назв. р-ции Норриша типа П, в отличие от р-ции Норриша
типа I, заключающейся в фотодиссоциации (предиссоциации) по связям, примыкающим
к карбонильной группе:
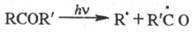
Образующийся в р-ции Норриша
типа П промежут. бирадикал может не только распадаться, но и циклизоваться,
приводя к образованию замещенного циклобутанола.
Широко распространены также
р-ции присоединения протона к таким основаниям (напр., к акридину), у к-рых
при переходе в возбужденное состояние значительно увеличивается основность:

Лит. см. при ст. Фотохимия. М.Г. Кузьмин.