


СОЛЬВАТАЦИЯ, взаимод.
молекул растворенного в-ва (или их ассоциатов) с молекулами р-рителя. Приводит
к изменению св-в молекул в р-ре (в сравнении со св-вами газовой фазы), влияет
на все физ. и физ.-хим. процессы, протекающие в р-рах, в т.ч. определяет скорость
реакций в растворах и положение равновесия, а в ряде случаев и их механизм.
С. в водных средах часто наз. гидратацией. Наиб. интенсивна С. ионов
в растворах электролитов.
С. состоит в том, что молекула
растворенного в-ва оказывается окруженной сольватной оболочкой, состоящей из
более или менее тесно связанных с ней молекул р-рителя. В результате С. образуются
сольваты-мол. образования постоянного или переменного состава. Время жизни соль-ватов
определяется характером и интенсивностью межмолекулярных взаимодействий;
даже в случае сильного взаимод. время жизни отдельного сольвата мало из-за
непрерывного обмена частицами в сольватной оболочке. В соответствии с типами
межмол. взаимод. выделяют неспецифическую и специфическую С. Неспецифическая
С. обусловлена ван-дер-ваальсовыми взаимод., специфическая С. проявляется гл.
обр. вследствие электростатич. взаимод., коор-динац. и водородных связей.
Важнейшие термодинамич.
характеристики С.-энталь-пия С. DHc и энергия Гиббса
С. (своб. энергия С.) DGc, связанные соотношением:
DGc=
DHc-ТDSc,
где DSc-энтропия
С., T-абс. т-ра. Энтальпия С. определяет тепловой эффект внедрения молекулы
растворенного в-ва в р-ритель; энергия Гиббса С. определяет растворимость
в-ва.
Наиб. простой способ эксперим.
определения энтальпии С. состоит в непосредств. измерении теплового эффекта
растворения в-ва А в р-рителе S-энтальпии растворения DHрА/S-и
использовании соотношения:
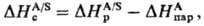
где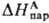 -энтальпия
парообразования в-ва А. Благодаря развитию
калориметрич. техники определение энтальпий растворения возможно практически
для всех систем; осн. проблема состоит в корректном определении энтальпий парообразования.
В то же время измерения значений DGc достаточно трудны,
особенно в случаях С. ионов в неводных р-рах. Нередко вместо DGc
вычисляют изменение этой величины D(DGc) относительно
ее значения в водной среде, используя для этого стандартную молярную энергию
DGп переноса иона X из воды W в к.-л. р-ритель S:
-энтальпия
парообразования в-ва А. Благодаря развитию
калориметрич. техники определение энтальпий растворения возможно практически
для всех систем; осн. проблема состоит в корректном определении энтальпий парообразования.
В то же время измерения значений DGc достаточно трудны,
особенно в случаях С. ионов в неводных р-рах. Нередко вместо DGc
вычисляют изменение этой величины D(DGc) относительно
ее значения в водной среде, используя для этого стандартную молярную энергию
DGп переноса иона X из воды W в к.-л. р-ритель S:
D(DGc)
= DGп(X, W:S)=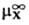 (в
р-рителе S)-
(в
р-рителе S)-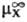 (в
воде),
(в
воде),
где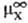 -стандартный
хим. потенциал иона X (рассматривается бесконечно разб. р-р).
-стандартный
хим. потенциал иона X (рассматривается бесконечно разб. р-р).
Структура ближайшего окружения
частицы растворенного в-ва характеризуется координационными числами С., определяемыми
как кол-во молекул р-рителя, связанных достаточно долго с этой частицей, чтобы
участвовать вместе с ней в диффузионном движении. Число С. зависит от природы
растворенной частицы и р-рителя, а также в нек-рой степени от используемого
метода определения; обычно используют данные по сжимаемости р-ра, скорости
диффузии ионов, электропроводности, а также термохим.
методы, электронное спиновое эхо и др. Для одновалентных ионов щелочных металлов
и галогенов числа С. составляют от 0,5 до 5,0 (значения меньше 1 свидетельствуют
о том, что в нек-рые моменты времени сольват-ная оболочка отсутствует).
В бинарных р-рителях, состоящих
из нейтрального (не-полярного) и активного (полярного) компонентов, возникает
селективная С., при к-рой состав сольватной оболочки резко отличается от состава
р-ра в целом. Особенно сильна селективная С. при малых концентрациях полярного
компонента.
При исследовании динамич.
поведения молекул в р-рах, их реакц. способности, для описания С. короткоживущих
состояний используют понятие неравновесной С. (неравновесной среды), при к-рой
состав и строение сольватных оболочек не отвечают минимуму своб. энергии системы,
достижимому при условии бесконечности времени жизни данных состояний. Напр.,
состояния молекул, из к-рых происходит оптич. (излучательный) квантовый переход,
всегда сольватированы неравновесно. Неравновесность среды определяется как вращательными,
так и трансляц. степенями свободы молекул р-рителя. Релтаксация среды к
равновесию происходит по закону ехр(-t/тL), где
t- время, тL- характеристика релаксац. способности
р-рителя. Для воды, напр., тL = 0,25·10-12
с.
Сольватирующая способность
р-рителя оценивается по ряду эмпирич. параметров с использованием эмпирич. шкал
р-рителей. Иногда пользуются понятием "сила р-рителя", основанным
на предположении о независимости сольвати-рующей способности р-рителя от св-в
растворяемого в-ва. Одной из наиб. универсальных характеристик сольвати-рующей
способности р-рителя является его диэлектрич. проницаемость e.
Впервые влияние р-рителя
на кинетику р-ций этерифика-ции было обнаружено М. Бертло в 1854; впоследствии
Н.А. Меншуткин установил (1890), что хим. р-цию нельзя рассматривать отдельно
от среды, в к-рой она протекает. Возможность теоретич. расчета влияния р-рителя
на реакц. способность и статич. св-ва молекул растворенного в-ва определяется
гл. обр. разработанностью теории жидкого состояния (см. Жидкость).
В рамках статистич. теории, являющейся основой совр. представлений о структурных
и энергетич. св-вах жидкостей и р-ров, полный потенциал F взаимод. молекулы
растворенного в-ва со средой, находящейся в термодинамич. равновесии, имеет
для одноцент-ровых частиц (напр., атомов благородных газов) вид:
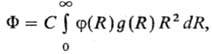
где R-расстояние
между частицами, f(R)-потенциал парного взаимод. молекул, g(R)-
радиальная корреляц. ф-ция распределения, С-постоянная, зависящая, в
частности, от плотности среды. Потенциал F позволяет определить энергию
межмол. взаимод., если известны ф-ции f(R) и g(R).
Применительно к изучению С. такой подход сопряжен с большими математич.
трудностями, т. к. не разработана общая теория, позволяющая с достаточной точностью
вычислять для реальных систем энергию межмол. взаимод. в широкой области изменения
R. Разработаны более простые, в т. ч. модельные, подходы к расчету DHc
и DGc, в частности макроскопич. (континуальные) и микроскопич.
(дискретные) способы описания эффектов С. Континуальные методы основаны на моделях
М. Борна, Л. Онсагера, Д. Кирквуда. Своб. энергия С. молекулы в среде равна:
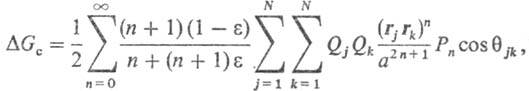
где а-радиус полости,
вырезаемой в результате внедрения молекулы растворенного в-ва в р-ритель, Qj,
Qk - эффективные заряды на j-м и k-м атомах
этой молекулы, N - число атомов
в ней, Рn-полиномы Лежандра, описывающие соотв. монопольные,
дипольные, октупольные взаимод. и эффекты более высоких порядков, Од-углы, образованные
векторами rj и rk, определяющими положения
атомов у и k. Частными случаями данного ур-ния являются ур-ния для своб.
энергии С. иона DG0-ур-ние Борна:
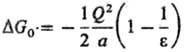
(Q-заряд иона) и
ур-ние Онсагера (модель реактивного поля):
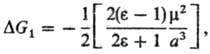
где m-дипольный момент
молекулы растворенного в-ва. Несмотря на широкое использование ур-ния Онсагера,
ряд опытных данных не подтверждается расчетом, напр. линейная зависимость энтальпии
и своб. энергии сольватации от дипольного момента m.
Более точные расчеты в
рамках микроскопии, подходов получены с использованием методов Монте-Карло и
мол, динамики. В методе мол. динамики с помощью ЭВМ численно решают классич.
ур-ния движения Ньютона, считая известной потенц. энергию взаимод. молекул.
Это позволяет "наблюдать" за движением отдельных молекул жидкости,
определять фазовые траектории, а затем усреднять их по времени и находить значения
требуемых термодинамич. и структурных ф-ций. Метод позволяет рассчитать статич.
и динамич. св-ва р-ров, в т. ч. и для неравновесных процессов. В методе Монте-Карло
состояния рассматриваемой системы частиц считаются случайными, задача же состоит
в отборе наиб. вероятных конфигураций и послед. усреднении по этим конфигурациям
разл. св-в. Ввиду этого метод приспособлен для расчета лишь равновесных величин.
Развитие ЭВМ позволяет применять оба метода ко все более широкому кругу объектов.
В результате оказывается возможным корректное разделение энтальпий и своб. энергий
С. на физически обоснованные вклады, связанные с разл. взаимод., и анализ зависимостей
между ними. Методы Монте-Карло и мол. динамики позволяют рассчитывать энтальпии
С. с точностью, сравнимой е экспериментальной (5-10 кДж/моль). Однако пока они
не позволяют учитывать взаимную поляризацию р-рителя и растворенного в-ва, а
также структурную перестройку в р-ре. Эти эффекты возможно определить с помощью
квантовохим. расчетов, к-рые позволяют прогнозировать строение и св-ва изолир.
молекул и механизмы р-ций, что необходимо для корректного выделения вклада,
обусловленного непосредственно влиянием р-рителя. Поверхности потенциальной
энергии молекул и реагирующих систем в газовой фазе и в р-рах могут иметь
принципиально разл. профиль.
С. приводит к тому, что
тип р-рителя изменяет скорость хим. р-ций (до 109 раз), определяет
относит. устойчивость таутомеров, конформеров, изомеров, влияет на механизм
р-ций. Положения кислотно-основных равновесий в значит. степени определяются
сольватирующей способностью р-рителя. Подробнее о влиянии С. на физ.-хим",
характеристики растворенных в-в и их реакц. способность см. в ст. Реакции
в растворах.
На влиянии С. на характеристики
электронных спектров поглощения и испускания основано явление, наз. сольватохромией.
Лит.: Бургер К.,
Сольватация, ионные реакции и комплексообразование в неводных средах, пер. с
англ., М., 1984; Симкин Б. Я." Шейхет И. И., Квантовохямичсская и статистическая
теория растворов. Вычислительные методы и их применение, М., 1989; Solvents
and solvent effects in organic chemistry, ed. by Ch. Reichardt, N.Y., 1988.
Б.Я. Симкин.