


НУКЛЕОФИЛЬНЫЕ РЕАКЦИИ,
гетеролитич. р-ции орг. соед. с нуклеоф. реагентами (нуклеофилами, от лат.
nucleus-ядро и греч. phileo-люблю). К н у к л е о ф и л а м относят анионы и
молекулы (орг. и неорг.), к-рые при участии в р-ции отдают свою пару электронов
на образование новой связи. Общая черта Н.р.-атака нуклеофила по электроно-дефицитному
центру, завершающаяся присоединением реагента или замещением уходящей группы,
напр. нуклеоф. замещение у насыщ. атома углерода или в ароматич. кольце, нуклеоф.
присоединение к карбонильной группе или алкенам и алкинам, нуклеоф. замещение
у карбонильного атома углерода, нуклеоф. замещение у атома фосфора.
Наиб. изучены р-ции н у
к л е о ф. з а м е щ е н и я у н а с ы щ. а т о м а у г л е р о д а:

Р-ции такого типа обычно
используют также для качеств. и количеств. определения понятий, характеризующих
Н.р.
В этих р-циях нуклеофилом
является частица X:, предоставляющая орг. субстрату пару электронов. Для уходящей
со своей парой электронов группы Z: принято назв. н у к л е о-ф у г (от лат.
nucleus-ядро и fugio-убегаю). На скорость и механизм р-ции нуклеоф. замещения
определяющее влияние оказывают нуклеоф. реакц. способность (или нуклеофильность,
"нуклеофильная сила") реагента X:, нуклеофугная рсакц. способность
(или нуклеофугность) уходящей группы Z:, природа субстрата и условия р-ции (т-ра,
р-ритель, давление и т. д.). Нуклеофильность, в отличие от основности, является
величиной кинетической, а не термодинамической, т. е. количеств. мерой нуклеоф.
реакц. способности служит константа скорости р-ции, а не константа равновесия.
Различают два предельных
случая р-ций нуклеоф. замещения-мономол. процесс SN1 и
бимолекулярный (синхронный) SN2:
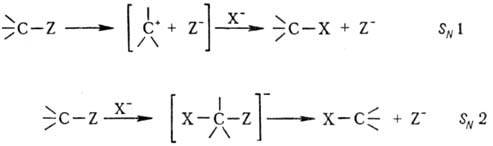
Механизм нуклеоф. замещения
существенно зависит от природы субстрата и р-рителя. Так, процессы SN1
реализуются в полярных р-рителях (Н2О, СН3ОН, АсОН
и др.), способствующих гетеролизу связи С—Z, и в р-циях с субстратами, содержащими
третичный, аллильный или бен-зильный атом С. Процессы SN2
в меньшей степени зависят от р-рителя и наиб. характерны для субстратов
с первичным атомом С.
При мономол. процессе первоначально
под действием р-рителя происходит ионизация субстрата с образованием трехкоординац.
карбкатиона и нуклеофуга (эта стадия обычно определяет скорость всего процесса),
а затем следует быстрая стадия связывания карбкатиона с нуклеофилом. При этом
атака нуклеофила равновероятна с обеих сторон, и в случае асимметрич. реакц.
центра обычно наблюдается образование рацематов. При бимол. процессе образуется
пятикоординац. переходное состояние, причем атака нуклеофила осуществляется
со стороны, противоположной уходящему заместителю, что приводит к обращению
конфигурации, напр. т. наз. вальденовское обращение (см. Динамическая стереохимия).
Р-ция SNl
обычно имеет суммарный 1-й порядок; скорость ее, как правило, не зависит
от природы нуклеофила и его концентрации, но сильно зависит от природы нуклеофуга
и р-рителя. Кинетика р-ции SN2 описывается ур-нием
2-го порядка - первого по субстрату и первого по нуклеофилу. Скорость р-ции
в этом случае зависит как от концентрации, так и от хим. природы нуклеофила.
Известно неск. подходов
к количеств. оценке нуклеоф. реакц. способности реагента X: на основе корреляц.
соотношений как в SN2-, так и SN1-процессах.
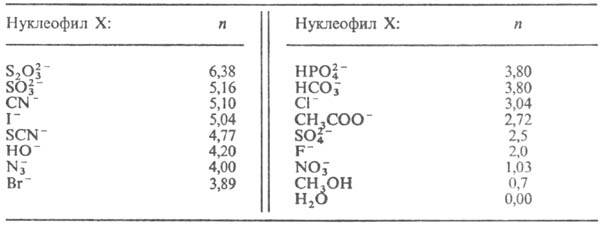
Для р-ций SN2 в воде или метаноле наиб. широко применяют
ур-ние Свена-Скотта lg (k/k0) = S.n,
где k и k0-константы скорости р-ции субстрата соотв.
с данным нуклеофилом и водой, S- параметр чувствительности субстрата
к изменению нуклеофила (S = 1 для стандартного субстрата-СН3Вr),
и-параметр нуклеофильности реагента (табл. 1).
Табл. 1.-ЗНАЧЕНИЕ
ПАРАМЕТРА НУКЛЕОФИЛЬНОСТИ п ДЛЯ НЕКОТОРЫХ РЕАГЕНТОВ (вода, 25 °С)
Для процессов типа SNl
справедливо корреляц. ур-ние Ритчи lg(k/k0) = N+.
Оно получено измерением скоростей р-ций с использованием в качестве субстратов
карбкатио-нов, стабилизированных арилъными заместителями три-фенилметанового
ряда.
Параметр нуклеофильности
N+ характеризует реакц. способность нуклеофила в определенном р-рителе;
в воде величины N+ близки к параметрам п.
Величины параметров нуклеофильности
могут заметно меняться в зависимости от конкретной Н.р., однако общая тенденция
изменения нуклеофильности обычно сохраняется. Так, практически во всех Н.р.
ОН-, CN-, RS-, I- и Вr-проявляют
себя как сильные нуклеофилы, а Н2О, СН3ОН, F-,
NO-3, SО42--как слабые.
Мерой нуклеофугности могут
служить константы скорости сольволиза (протекающего по механизму SN1)
однотипных субстратов, отличающихся лишь природой уходящей группы (табл.
2).
Табл. 2. ОТНОСИТЕЛЬНЫЕ
КОНСТАНТЫ СКОРОСТИ СОЛЬВОЛИЗА (kотн)
НЕКОТОРЫХ УХОДЯЩИХ ГРУПП [субстрат-Ph(CH3)CHZ в 80%-ном водном
этаноле, 75°С]

К "хорошим"
нуклеофугам относят орг. сульфонат-(този-лат, мезилат, трифлат), фторсульфат-(FSО-3)
и перхлорат-анионы(СlO-4). Ковалентные орг. производные
этих анионов широко используют в качестве алкилирующих реагентов-чрезвычайно
активных субстратов в Н.р. Еще более хорошие нуклеофугные частицы-азот из алкилдиазониевых
солей (RN+2), трехвалентный иод (напр., группа IСl2),
вода из протонир.
спирта  и простой эфир из триалкилоксониевых
солей
и простой эфир из триалкилоксониевых
солей  ;
однако алифатич. субстраты, содержащие в своей структуре эти группы, при комнатной
т-ре обычно неустойчивы и используются лишь в качестве активных интермедиатов,
генерируемых непосредственно в реакц. среде.
;
однако алифатич. субстраты, содержащие в своей структуре эти группы, при комнатной
т-ре обычно неустойчивы и используются лишь в качестве активных интермедиатов,
генерируемых непосредственно в реакц. среде.
Существует неск. разл.
подходов к теоретич. интерпретации понятий нуклеофильности и нуклеофугности
и к оценке факторов, влияющих на их величину. Осн. факторы - основность (кислотность),
поляризуемость, сольватац. эффекты, величины потенциалов ионизации и окисления,
стерич. и электростатич. эффекты, наличие своб. электронной пары у атома, соседнего
с нуклеоф. центром, прочность связи с атомом углерода. Следует отметить, что
прямой корреляции нуклеофильности с к.-л. одним из этих параметров обычно нет,
как нет и корреляции между нуклеофильностью и нуклеофугностью; напр., тиолят-анион
RS--хороший нуклеофил, но слабое основание и "плохая"
уходящая группа, гидроксид-анион НО- -хорошее основание и нуклеофил,
но плохая уходящая группа. Анионы самых сильных к-т-хлорной и трифторметансульфоновой
- хорошие нуклеофу-ги и в то же время способны проявлять нуклеофильные свойства.
Нуклеоф. замещение в алифатич.
ряду имеет исключительно важное значение для орг. синтеза, позволяя целенаправленно
заменять функц. группы, а также конструировать углеродный скелет молекулы путем
использования С-нуклеофилов (напр., металлоорг. соединений).
Н у к л е о ф. з а м е
щ е н и е в а р о м а т и ч. р я д у (ароматич. нуклеоф. замещение) обычно сильно
затруднено и может протекать по механизмам "присоединение - отщепление"
или через промежут.
образование дегидробензола (арино-вьш механизм):
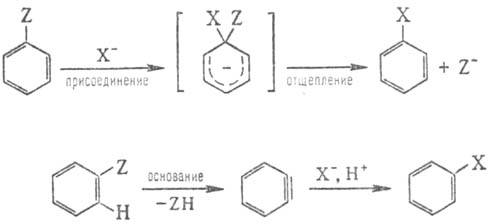
В первом случае первоначально
происходит присоединение нуклеофила к ароматич. субстрату с образованием промежут.
продукта (иногда стабильного - т. наз. комплекс Майзенхаймера), отщепление нуклсофуга
от к-рого приводит к конечному продукту замещения. Электроноакцеп-торные заместители
в ядре (NO2, COR, CN и др.) стабилизируют интермедиат и способствуют
быстрому протеканию р-ции. Так, если галоген в хлорбензоле можно заместить действием
щелочи в очень жестких условиях (400 °С; 4,5 МПа), то 2,4-динитрохлорбензол
и 2,4,6-тринитрохлор-бензол реагируют с нуклеоф. реагентами при комнатной т-ре
или умеренном нагревании.
Ариновый механизм обычно
реализуется в р-циях арил-галогенидов с сильными основаниями, напр. NaNH2
в NH3.
Очень важны для орг. синтеза
р-ции н у к л е о ф. п р ис о е д и н е н и я и з а м е щ е н и я у к а р б
о н и л ь н о г о а
т о м а у г л е р о д а:
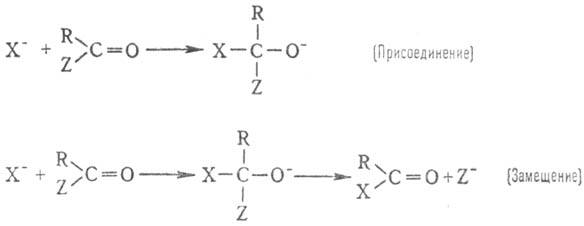
В нек-рых случаях процесс
останавливается на стадии присоединения, напр. в р-циях HCN с альдегидами и
кетонами с получением циангидринов. В др. случаях (обычно когда Z: хороший нуклеофуг,
напр. галоген или вода) образуются продукты замещения. К этому типу процессов
относят практически важные р-ции этерификации (эти р-ции протекают при наличии
кислотного катализа): RCOOH + + R'OH RCOOR'
+ Н2О, а также р-ции карбонильных производных с С-нуклеофилами, приводящие
к возникновению новой связи углерод-углерод.
RCOOR'
+ Н2О, а также р-ции карбонильных производных с С-нуклеофилами, приводящие
к возникновению новой связи углерод-углерод.
Н у к л е о ф. п р и с
о е д и н е н и е к к р а т н о й с в я з и у
г л е р о д-у г л е р о д обычно возможно лишь при наличии в молекуле субстрата
электроноакцепторных заместителей. Примером может служить присоединение нуклеоф.
реагентов (напр., воды) к a,b-непредсльным к-там: СН2=СНСООН + Н2О НОСН2СН2СООН.
К частному случаю нуклеоф. присоединения к двойной связи можно отнести очень
важную для орг. синтеза Михаэля реакцию.
НОСН2СН2СООН.
К частному случаю нуклеоф. присоединения к двойной связи можно отнести очень
важную для орг. синтеза Михаэля реакцию.
Лит.: Хадсон Р.
Ф., в кн.: Реакционная способность и пути реакций, пер. с апгл., М., 1977, с.
175-261; Кери Ф., Сандберг Р., Углубленный курс органической химии, пер. с англ.,
М., 1981; Марч Дж., Органическая химия, пер. с англ., М., 1987, т. 2, с. 11-243;
там же, т. 3, с. 5-45; Ibne-Rasa K.M., "J. Chem. Educ.", 1967, v.
44, № 2, p. 89-94; Stirling C.J.M., "Ace. Chem. Res.", 1979, v.
12, p. 198-203; Harris J.M., McManusS.P., Nucleophilicity, Wash., 1987.
В. В. Жданкин.