


МОНОМОЛЕКУЛЯРНЫЕ РЕАКЦИИ,
элементарные хим. р-ции, в к-рых изменяется состав или строение только одной
молекулы, радикала или иона, напр. распад, изомеризация. Часто М.р., особенно
распад молекулы (обычно на два фрагмента), являются начальными стадиями сложных
процессов - крекинга, деструкции полимеров и др. В газах при повышении т-ры
Т или уменьшении плотности хим. равновесие смещается в сторону продуктов
распада, вследствие чего значительно возрастают теплоемкость и энтальпия системы.
Константы равновесия М. р. необходимы для расчета равновесного состава и термодинамич.
ф-ций газов и приводятся в справочниках.
М.р., как правило, эндотермичны,
и для р-ции необходима активация - переход частицы А в активное состояние А*,
энергия к-рого достаточна для преодоления потенц. барьера на пути р-ции. Активная
молекула может самопроизвольно превратиться в продукт либо дезактивироваться.
Эти качеств. черты М.р. описываются схемой Лин-демана:
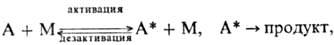
где М-частицы среды (атомы,
молекулы, электроны, ионы) или стенка сосуда. Стадия самопроизвольного распада
в более детальной схеме (Р.А. Маркус, O.K. Раис) представляется двумя последоват.
переходами: 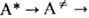 продукт, где
продукт, где 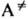 -переходное
состояние (активир. комплекс), в к-ром энергия, достаточная для р-ции, уже сосредоточена
на разрываемой связи (в общем случае-на координате реакции).
-переходное
состояние (активир. комплекс), в к-ром энергия, достаточная для р-ции, уже сосредоточена
на разрываемой связи (в общем случае-на координате реакции).
Константа скорости М.р.
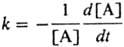 , где
[А]-концентрация реагирующих молекул, t-время. При заданной т-ре k
пропорциональна давлению р в области низких р (обозначается
k0) и не зависит от р в области достаточно высоких
р (обозначается
, где
[А]-концентрация реагирующих молекул, t-время. При заданной т-ре k
пропорциональна давлению р в области низких р (обозначается
k0) и не зависит от р в области достаточно высоких
р (обозначается 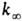 ).
В переходной области давлений k монотонно изменяется от k0
до
).
В переходной области давлений k монотонно изменяется от k0
до 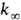 . Для
молекул, состоящих из 5-10 атомов, при ~ 300-1000 К середина переходной области,
определяемая условием k/
. Для
молекул, состоящих из 5-10 атомов, при ~ 300-1000 К середина переходной области,
определяемая условием k/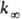 = 1/2, расположена обычно при р ~ 102-104 Па.
С уменьшением числа атомов в молекуле и с повышением Т переходная область
смещается в сторону больших р. В рамках схемы Линдемана зависимость k
от р объясняется тем, что при низких р лимитирующая стадия
р-ции - активация, имеющая второй порядок, а при высоких р-стадия А*
= 1/2, расположена обычно при р ~ 102-104 Па.
С уменьшением числа атомов в молекуле и с повышением Т переходная область
смещается в сторону больших р. В рамках схемы Линдемана зависимость k
от р объясняется тем, что при низких р лимитирующая стадия
р-ции - активация, имеющая второй порядок, а при высоких р-стадия А*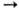 продукт,
имеющая первый порядок. В двухатомных молекулах имеется всего одна колебат.
степень свободы, и колебания всегда соответствуют движению вдоль координаты
р-ции. Состояния А* и
продукт,
имеющая первый порядок. В двухатомных молекулах имеется всего одна колебат.
степень свободы, и колебания всегда соответствуют движению вдоль координаты
р-ции. Состояния А* и 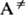 двухатомной молекулы тождественны, самопроизвольный распад происходит за время
порядка периода колебаний. Это время практически всегда настолько мало, что
ниж. граница переходной области давлений соответствует очень большим плотностям,
не реализуемым в газах. Поэтому диссоциация двухатомных молекул в газах всегда
имеет второй порядок. Для М.р. в конденсир. средах k практически не зависит
от р.
двухатомной молекулы тождественны, самопроизвольный распад происходит за время
порядка периода колебаний. Это время практически всегда настолько мало, что
ниж. граница переходной области давлений соответствует очень большим плотностям,
не реализуемым в газах. Поэтому диссоциация двухатомных молекул в газах всегда
имеет второй порядок. Для М.р. в конденсир. средах k практически не зависит
от р.
Наблюдаемая зависимость
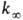 от Т
приближенно описывается ур-нием Аррениуса с энергией активации
от Т
приближенно описывается ур-нием Аррениуса с энергией активации 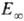 ,
к-рая для распада обычно мало отличается от энергии D разрываемых связей,
и предэкспоненц. множителем А, значение к-рого составляет в большинстве
случаев 1013-1015 с-1. Большие значения А
могут соответствовать случаям, когда при переходе
,
к-рая для распада обычно мало отличается от энергии D разрываемых связей,
и предэкспоненц. множителем А, значение к-рого составляет в большинстве
случаев 1013-1015 с-1. Большие значения А
могут соответствовать случаям, когда при переходе 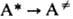 в молекуле появляются новые вращат. степени свободы. Существ. отклонения
в молекуле появляются новые вращат. степени свободы. Существ. отклонения 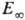 от D и аномально малые значения А могут наблюдаться, если р-ция
идет по т. наз. н е а д и а б а т и ч е с к о м у к а н а л у (т.е.
с изменением электронного состояния молекул). С повышением
т-ры адиабатич. канал протекания р-ции (без изменения электронного состояния
молекул) дает больший вклад в суммарное значение константы скорости. В той области
т-р, где протекание р-ции по обоим каналам дает сопоставимые вклады, константу
скорости k выражают в виде суммы двух ур-ний Аррениуса-с двумя значениями
энергии активации и предэкспоненц. множителя. Зависимость k0
от Т выражена слабее, чем
от D и аномально малые значения А могут наблюдаться, если р-ция
идет по т. наз. н е а д и а б а т и ч е с к о м у к а н а л у (т.е.
с изменением электронного состояния молекул). С повышением
т-ры адиабатич. канал протекания р-ции (без изменения электронного состояния
молекул) дает больший вклад в суммарное значение константы скорости. В той области
т-р, где протекание р-ции по обоим каналам дает сопоставимые вклады, константу
скорости k выражают в виде суммы двух ур-ний Аррениуса-с двумя значениями
энергии активации и предэкспоненц. множителя. Зависимость k0
от Т выражена слабее, чем 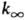 .
.
Р-ция, обратная мономолекулярному
распаду, является бимолекулярной и имеет третий (при низких р)или второй
(при высоких р)порядок. Изомеризация является М. р. как в прямом, так
и в обратном направлении.
Существует неск. теоретич.
представлений, на основании к-рых возможен приближенный расчет константы скорости
М. р. Для адиабатич. р-ций наиб. удовлетворит. результаты дает статистич. т
е о р и я Р а и с а-Р а м с п е р г е р а-К а с с е-л я-М а р к у с а (сокращенно
РРКМ). В этой теории предполагается, что заселенности всех квантовых состояний
молекулы с одинаковой энергией одинаковы независимо от различий в скорости возникновения
и гибели каждого состояния. Это позволяет найти ф-цию распределения молекул
по энергии f(E), к-рая содержит лишь усредненные константы скорости
перехода из состояния с энергией Е' в состояние с энергией Е [обозначается
k(E, E')] и самопроизвольного распада [обозначается k(E)].
Ф-ция k(E)вычисляется на основе активированного комплекса теории
и выражается соотношением:
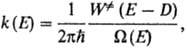
где 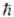 -
постоянная Планка,
-
постоянная Планка, 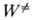 (Е
— D) - число состояний активир. комплекса с энергией в интервале значений
от нуля до (Е — D), причем учитываются все состояния, кроме относящихся
к координате р-ции, W (E)-плотность колебат. и вращат. состояний, т.е.
число состояний молекулы, приходящихся на единичный интервал энергии вблизи
Е. Ф-ция k(E, E')определяется механизмом передачи энергии при
столкновениях. Если вероятность дезактивации при одном столкновении близка к
единице (т. наз. с и л ь н ы е с т о л к н ов е н и я), эта ф-ция имеет вид:
(Е
— D) - число состояний активир. комплекса с энергией в интервале значений
от нуля до (Е — D), причем учитываются все состояния, кроме относящихся
к координате р-ции, W (E)-плотность колебат. и вращат. состояний, т.е.
число состояний молекулы, приходящихся на единичный интервал энергии вблизи
Е. Ф-ция k(E, E')определяется механизмом передачи энергии при
столкновениях. Если вероятность дезактивации при одном столкновении близка к
единице (т. наз. с и л ь н ы е с т о л к н ов е н и я), эта ф-ция имеет вид:
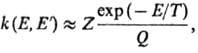
где Q-статистич.
сумма, Z- число столкновений в единицу времени (газокинетич. число).
Расчет f(E), k, 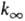 и
k0 производят по ф-лам:
и
k0 производят по ф-лам:
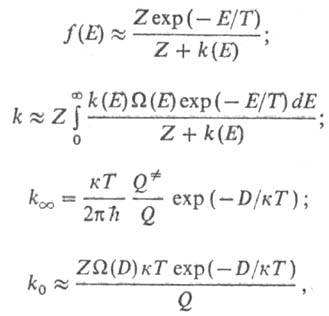
где к -постоянная
Больцмана, 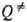 -статистич.
сумма активир. комплекса. Результаты вычислений k0 для др.
механизмов активации интерпретируют посредством замены Z на PZ, где р-коэф.,
определяемый механизмом активации и т-рой. Выражение для
-статистич.
сумма активир. комплекса. Результаты вычислений k0 для др.
механизмов активации интерпретируют посредством замены Z на PZ, где р-коэф.,
определяемый механизмом активации и т-рой. Выражение для 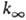 не зависит от механизма активации.
не зависит от механизма активации.
Константы 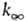 и k0 вычисляют с точностью в среднем до множителя 3-10. Погрешность
в осн. связана с неопределенностью исходных данных о k (Е, Е')и
и k0 вычисляют с точностью в среднем до множителя 3-10. Погрешность
в осн. связана с неопределенностью исходных данных о k (Е, Е')и 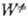 (Е
— D), ведущей к погрешностям вычислений соотв. b и
(Е
— D), ведущей к погрешностям вычислений соотв. b и 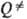 .
Удовлетворит. результаты удается получить путем коррекции множителей b и
.
Удовлетворит. результаты удается получить путем коррекции множителей b и 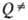 по эксперим. данным с о0 и
по эксперим. данным с о0 и 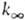 при
одном значении т-ры (или в узком интервале т-р). Сравнение результатов вычислений
с экспериментом показывает, что р слабо уменьшается с возрастанием т-ры, а при
~ 1000 К, как правило, по порядку величины р составляет 0,1. Это свидетельствует
о значит. отклонении механизма активации от механизма сильных столкновений.
при
одном значении т-ры (или в узком интервале т-р). Сравнение результатов вычислений
с экспериментом показывает, что р слабо уменьшается с возрастанием т-ры, а при
~ 1000 К, как правило, по порядку величины р составляет 0,1. Это свидетельствует
о значит. отклонении механизма активации от механизма сильных столкновений.
Дальнейшее развитие теории
связано с исследованиями возможных динамич. ограничений, налагаемых на перераспределение
энергии и на скорость самопроизвольного превращ. активной молекулы, более точным
количеств. определением k(E,E')на основе эксперим. данных об эффективных
сечениях передачи энергии при столкновениях или квантовомех. расчетов. Наряду
с аналит. подходами к решению этих вопросов быстро развиваются методы численного
моделирования на ЭВМ процессов внутримолекулярного движения, активации и дезактивации.
Как правило, моделирование проводится в рамках классич. механики.
Совр. практика ставит и
принципиально новые задачи-изучение кинетики М. р. в неравновесных условиях
или при неравновероятном распределении заданной энергии по разл. группам степеней
свободы реагирующей молекулы. Такие условия реализуются, напр., при избират.
воздействии внеш. источников энергии на нeк-рые степени свободы молекулы (возбуждение
колебат. состояний молекулы электронным ударом
в плазме электрич. разряда, ИК излучением лазера и т.д.) в быстрых газодинамич.
процессах (сжатие в ударных волнах, истечение газа из сопла в разреженное пространство
и др.). Подобные задачи не м. б. решены в рамках статистич. теории и требуют
более детального изучения в кинетике М.р. роли разл. видов внутримолекулярного
движения (колебаний определенного типа, вращений, электронного возбуждения)
и разл. процессов передачи энергии при столкновениях. В ряде случаев кинетику
М.р. удается исследовать в рамках обобщенной статистич. теории путем представления
неравновесной среды или системы колебат. и вращат. степеней свободы молекулы
в виде неск. подсистем, каждая из к-рых характеризуется своим значением т-ры
(см. Неравновесная химическая кинетика).
Лит.: Робинсон П.,
Холбрук К., Мономолекулярные реакции, пер. с англ., М., 1975; Кузнецов Н. М.,
Кинетика мономолежуллрных реакций, М., 1982. Я. М. Кузнецов.