МАКРОМОЛЕКУЛА (от греч. makros - большой и молекула), молекула полимера. М. имеют цепное строение; состоят из одинаковых или разл. структурных единиц - составных звеньев, представляющих собой атомы или группы атомов, соединенные друг с другом ковалентными связями в линейные последовательности. Последовательность соединенных друг с другом атомов, образующих собственно цепь, наз. хребтом цепи, или цепью главных валентностей, а заместители у этих атомов - боковыми группами. М. могут иметь линейное или разветвленное строение; в разветвленных М. различают основную и боковые цепи. См. также Высокомолекулярные соединения.
Осн. мол. характеристики М. - хим. строение, длина цепи (степень полимеризации, относит. молекулярная масса) и гибкость.
Хим. строение звеньев и их взаимное расположение в цепи характеризуют первичную структуру М. Первичная структура исчерпывающе определяется конфигурацией М. - пространств. расположением атомов в М., к-рое не м. б. изменено без разрыва связей и обусловлено длинами связей и величинами валентных углов. Число разл. способов взаимного расположения (чередования) звеньев в М. характеризуется конфигурационной энтропией и отражает меру информации, к-рую может содержать М. Способность к хранению информации - одна из самых важных характеристик М., значение к-рой стало понятно после открытия генетич. кода и расшифровки структуры основных биол. М. - нуклеиновых кислот и белков.
Первичная структура синтетич. М. предопределяет (вместе с молекулярно-массовым распределением, т.к. реальные синтетич. полимеры состоят из М. разной длины) способность полимеров кристаллизоваться, быть каучуками, волокнами, стеклами и т. п., проявлять ионо- или электронообменные св-ва, быть хемомех. системами (т.е. обладать способностью перерабатывать хим. энергию в механическую и наоборот). С первичной структурой связана также способность М. к образованию вторичных структур (см. ниже). В биополимерах, состоящих из строго идентичных М., эти структуры достигают высокой степени совершенства и специфичности, предопределяя способность, напр., белков быть ферментами, переносчиками кислорода и т.п.
М. способны к изменению формы и линейных размеров в результате теплового движения, а именно - ограниченного вращения звеньев вокруг валентных связей (внутр. вращение) и связанного с ним изменения конформации М., т. е. взаимного расположения в пространстве атомов и групп атомов, соединенных в цепь, при неизменной конфигурации М. Обычно в результате такого движения М. приобретает
наиб. вероятную форму статистич. клубка. Наряду с беспорядочной конформацией статистич. клубка могут существовать упорядоченные (спиральные, складчатые) конформации, к-рые обычно стабилизированы силами внутри-и межмол. взаимодействия (напр., водородными связями). В результате внутримол. взаимодействия м.б. получены М. в предельно свернутой конформации, наз. глобулой.
Ограничения внутр. вращения количественно описываются в терминах поворотной изомерии (см. Внутреннее вращение молекул). Для фрагмента М., построенной из атомов углерода, соединенных простыми связями, схема энергетич. барьеров внутр. вращения изображена на рисунке. Степень свободы этого вращения определяет гибкость М., с к-рой связаны каучукоподобная эластичность, способность полимеров к образованию надмолекулярных структур, почти все их физ. и мех. св-ва. Разница энергии Де между минимумами на кривой зависимости внутр. энергии Е от угла вращения j определяет термодинамич. (статич.) гибкость М., т.е. вероятность реализации тех или иных конформации (напр., вытянутых, свернутых), размер и форму М.; величины энергетич. барьеров DE определяют кинетич. (динамич.) гибкость М., т.е. скорость перехода из одной конформации в другую. Величины энергетич. барьеров зависят от размеров и характера боковых радикалов при атомах, образующих хребет цепи. Чем массивнее эти радикалы, тем выше барьеры. Конформация М. может изменяться и под действием внеш. силы (напр., растягивающей); податливость М. к таким деформациям характеризуется кинетич. гибкостью. При очень малых гибкостях, напр. в случаях лестничных полимеров или наличия действующей вдоль цепи системы водородных или координац. связей (см. Координационные полимеры), внутр. вращение сводится к относительно малым крутильным колебаниям мономерных звеньев друг относительно друга, чему соответствует макроскопич. модель упругой плоской лепты или стержня. Число возможных конформации М. возрастает с увеличением степени полимеризации, и термодинамич. гибкость по-разному проявляется на коротких и длинных участках М. Это можно понять с помощью др. макроскопич. модели - металлич. проволоки. Длинную проволоку можно скрутить в клубок, а короткую, у к-рой длина и размер в поперечном направлении соизмеримы, - невозможно, хотя физ. ее св-ва те же. Непосредств. численная мера термодинамич. гибкости (персистентная длина l) определяется выражением: l = l0exp(De/kT), где De > 0, l0 ~ 10-10 м (т.е. порядка длины хим. связи), k - постоянная Больцмана, Т - т-ра. Если контурная длина, т. е. длина полностью вытянутой М. без искажения валентных углов и связей, равна L, то L < l соответствует ситуации с короткой проволокой, и гибкость просто не может проявляться из-за малого числа допустимых конформации. При L >> l М. сворачивается в статистич. клубок, среднеквадратичное расстояние между концами к-рого <h2>1/2 при отсутствии возмущающих факторов пропорционально Р1/2 (Р - степень полимеризации).
Возмущающим фактором м. б. термодинамич. взаимодействие с р-рителем. Суть его удобно понять с помощью третьей модели - осмотической. М. как малая система из Р связанных в цепь элементов (мономерных звеньев) в своем поведении примерно эквивалентна системе из тех же Р элементов, но не связанных между собой, а заключенных в гибкую оболочку, не проницаемую для этих элементов и проницаемую для р-рителя. Такая система по сравнению с системой в идеальном р-рителе увеличит из-за осмотич. набухания свои линейные размеры в a раз. То же происходит и с линейными размерами клубка, в к-рый свернута М. Они становятся равными <h02>1/2a, где индекс "0" соответствует идеальному р-рителю. Для идеального р-рителя (Q - р-ритель) второй вириальный коэф. осмотич. ур-ния равен нулю, а a = 1. Т-ра, при к-рой a = 1, наз. тета (Q)-температурой Флори; a - параметр набухания Флори; он зависит от Рg (g [ 0,1). См. Набухание полимеров, Растворы полимеров.
Способность М. к образованию статистич. клубка является непосредств. проявлением термодинамич. гибкости, а размеры клубка <h02>1/2 служат количеств. характеристикой этой гибкости. Иногда вместо l пользуются величиной статистич. сегмента Куна А. При этом моделью М. служит эквивалентная цепь, состоящая из статистич. сегментов длиной А, абсолютно свободно вращающихся друг относительно друга. Величина А пропорциональна числу звеньев в сегменте п, при к-ром полностью утрачивается корреляция между направлениями первого и n-го звеньев сегмента. Величина сегмента Куна также служит мерой гибкости М. и связана с персистентной длиной простым соотношением: А = 2l.
Для ряда теоретич. выкладок и практич. расчетов удобно пользоваться др. параметром - степенью развернутости клубка: b = <h02>1/2/L. Величина b = 0,25, соответствующая числу статистич. сегментов в М. А ~ 13, определяет границу между олигомерами и полимерами. При b < 0,25 М. образует свернутый клубок. Поскольку в реальных полимерах длина (размер) мономерных звеньев может варьировать в широких пределах, степень полимеризации сегмента Куна x является более информативной мерой гибкости, чем длина А (или l). На основании оценок х линейные полимеры относят к гибкоцепным, полужестким или жесткоцепным. Гибкоцепными полимерами принято считать те, у к-рых x не превышает 15. Условная граница между гибкоцепными и полужесткими М. лежит в области х ~ 15-20.
По аналогии с персистентной длиной l можно ввести характеристич. время: t = t0exp(DE/kT). Если абс. высота энергетич. потенц. барьеров, разделяющих поворотные изомеры (см. рис.), DE ~ kT, гибкость успевает проявиться за время t0 ~ 10-11 с как в покое, так и при приложении напряжения растяжения к р-ру или расплаву, т. е. т является мерой кинетич. гибкости. При DE >> kT удовлетворяется записанный выше экспоненциальный закон, но t можно значительно сократить приложением сильных мех. или гидродинамич. полей. Если DE > kT, но сопоставимо с kT, то время перехода одной конформации М. в другую определяется фундам. временем релаксации М.:
t = B.(Vh0/kT) (В - константа, V - объем М., h0 - вязкость р-рителя).
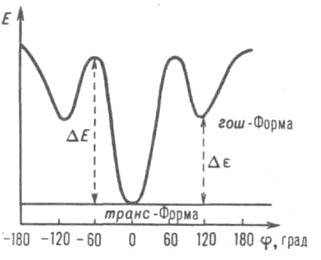 Зависимость внутр. энергии Е поворотных изомеров от угла вращения j [к определению термодинамич. (равновесной) и кинетич. (динамич.) гибкости].
Зависимость внутр. энергии Е поворотных изомеров от угла вращения j [к определению термодинамич. (равновесной) и кинетич. (динамич.) гибкости].
Зависимости размеров М. от Р описываются асимптотич. (Р : :) степенными соотношениями, подчиняющимися принципу масштабной инвариантности (скейлингу), и в общем случае имеют вид: <h02>1/2 ~ Pv (n - скейлинговый показатель). При n = 1/3 М. гибкая и имеет глобулярную конформацию; для М. в Q-р-рителе n = 1/2 ([h] ~ P1/2), в хорошем р-рителе n = 0,6 ([h] ~ P0,8); для полужестких М. ("протекаемых", т.е. р-ритель протекает через клубок) n = 2/3 ([h] ~ P), для жестких М. ("стержней") n = 1 ([h] ~ P1,7).
Физ. смысл скейлинговых показателей - характеристика заполненности координационной сферы М. (усредненный по времени и пространств. координатам объем,
занимаемый М., претерпевающей внутр. и внеш. тепловое движение) ее собственными звеньями. В набухшем клубке (хороший р-ритель) концентрация собственных звеньев составляет примерно 0,1%, в невозмущенном клубке (т.е. в Q-условиях) - от 1 до 3%, в глобуле - от 10 до 100%.
Гибкоцепные полимеры с ростом концентрации полимера в любом р-рителе стремятся к состоянию, когда размер М. пропорционален Р1/2. Напротив, М. жесткоцепных полимеров (x / 20) разворачиваются (n : 1,7), и возникает лиотропная жидкокристаллич. фаза. Переход в глобулярное состояние можно рассматривать как выпадение М. "на себя" (свертывание) при резком ухудшении качества р-рителя. Для глобул характерен только показатель n = 1/3, хотя степень заполненности клубка звеньями М. может быть намного ниже плотности полимера в блочном состоянии. Достаточным признаком глобулярного состояния является независимость характеристич. вязкости [h] от Р в данном р-рителе.
Линейные М. сложного строения способны к образованию вторичных структур (упорядоченное состояние М., возникающее в результате специфич. меж- и внутримолекулярных взаимод.). Это возможно обычно из-за дифильности и способности к избирательным взаимод. отдельных групп, входящих в М., между собой или с р-рителем. Вторичные структуры условно можно подразделить на линейно-кристаллич., жидкокристаллич., конденсац. и вулканизационные. Наиб. хорошо известным примером первых являются a-спирали в полипептидах, двойные спирали в нуклеиновых к-тах, тройные спирали в нек-рых полипептидах или фибриллярных белках. Принято считать, что соответствующие переходы типа спираль - клубок являются не фазовыми, а кооперативными. Жидкокристаллич. структуры возникают в гребнеобразных полимерах с мезогенными группами на концах ветвей (см. Жидкие кристаллы); малая мезофаза претерпевает почти те же фазовые переходы, что и ее низкомол. аналоги. Глобулы относят к категории конденсац. структур, но м. б. и такие случаи, когда в блоксополимерах отдельные блоки термодинамически несовместимы, происходит их сегрегация и в р-ре образуются мол. мицеллы с глобулярным ядром и рыхлой оболочкой типа клубка. Вулканизац. структуры чаще всего возникают в сополимерах, содержащих доноры и акцепторы протонов; при этом внутр. сетка образуется вследствие возникновения водородных связей между далеко (вдоль развернутой цепи) расположенными звеньями. С повышением густоты такой сетки постепенно происходит переход к глобулярному состоянию.
Лит.: Френкель С. Я., в сб.: Физика сегодня и завтра, Л., 1973, с. 176-270; его же, в кн.: Энциклопедия полимеров, т. 2, М., 1974, стлб. 100-33; Рафиков С. Р., Будтов В. П., Монаков Ю. Б., Введение в физико-химию растворов полимеров, М., 1978; Жен П. Ж. де, Идеи скейлинга в физике полимеров, пер. с англ., М., 1982; Эфрос А. Л., Физика и геометрия беспорядка, М., 1982; Гросберг А. Ю., Хохлов А. Р., Физика цепных молекул, М., 1984 (Новое в жизни, науке, технике. Сер. Физика, в. 8).
В. Г. Баранов. С. Я. Френкель.


