


КОРРЕЛЯЦИОННЫЕ СООТНОШЕНИЯ, характеризуют эмпирически устанавливаемые зависимости между разл. св-вами одного ряда хим. соед. (реакц. способностью, физ., термодинамич. св-ми, биол. активностью и др.) и параметрами структуры, среды или св-вами того же или др. ряда соединений. Формальный прототип большинства К. с. - ур-ниe Брёнстеда, выражающее найденную экспериментально линейную зависимость между константами скорости р-ций kc кислотного (основного) катализа и константами равновесия диссоциации к-ты (основания) Кa:
lgke=algKa+c, (1)
где а и с - эмпирич. параметры для данной р-ции. Ур-ние (1) - пример выполнения общего соотношения линейности своб. энергий (ЛСЭ), согласно к-рому изменения в своб. энергиях р-ций DG° и в своб. энергиях активации (DG++), вызываемые в разл. р-циях одинаковыми вариациями структуры реагирующих соед. или среды, связаны линейными зависимостями:
dRDGi=adRDGj+b, (2)
где dR - оператор изменения структуры (напр., при введении заместителя R) или среды, i, j - индексы р-ций. Ур-ние (2) отражает широкую эмпирич. закономерность: сходные изменения в строении соед. приводят к сходным изменениям в реакц. способности. Оно не вытекает из принципов термодинамики, поэтому К. с., основанные на соотношении ЛСЭ, принято относить к экстратермодинамическим. Важнейшее К. с. этого типа - ур-ние Гаммета, сопоставляющее изменения в константах скорости или равновесия, индуцируемые введением заместителей в ароматич. бензольное ядро в соед. I, с изменениями в константах диссоциации бензойных к-т II при аналогичном замещении:
dRDGi=аdRDG0БK, (3)
где DGi - своб. энергии р-ции или активации произвольной р-ции соед. I в произвольных (но одинаковых для данной серии-,соед.) условиях, а DG0БК - своб. энергия диссоциации бензойной к-ты в заданных (не обязательно идентичных) условиях. Ур-ние (3) сводится к виду:
lg(k/k0)=rlg(K/K0)БК (4)
где k - константа скорости (равновесия) р-ции соед. I с заместителем R, а k0 - та же константа для соед. с R=Н, К и К0 - константы диссоциации соответствующих бензойных к-т II, r - константа, характеризующая относительную (в сравнении с эталонной серией р-ций диссоциации бензойных к-т) чувствительность р-ций соед. I к структурным изменениям.
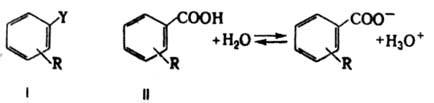
Реакц. серия диссоциации бензойных к-т в воде при 25 °С выбрана как стандартная, при этом величины lg(K/K0)=s представляют константы заместителей R. Ур-ние (4) известно как ур-ние Гаммета и обычно записывается в форме:
lgk=rs+lgk0 (5)
Ур-ние (5) позволяет с точностью b15% рассчитывать кинетич. и равновесные параметры мн. р-ций производных бензола с мета- и пaра-заместителями, располагая константами заместителей и величинами r, определенными статистически достоверно для неск. соед. данной реакц. серии. К. с. для структурных вариаций субстрата. В табл. приведены значения констант заместителей s для нек-рых наиб. важных групп (sм и sn - константы заместителей в мета- и пара-положениях). Для р-ций, ускоряемых электронодонорными заместителями (s<0), r<0, для р-ций, ускоряемых электроноакцепторными заместителями
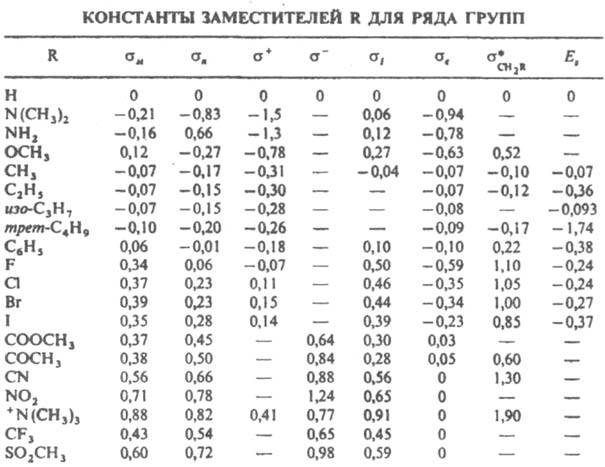
(s>0), r>0. Абс. величина r тем больше, чем более полярно переходное состояние лимитирующей стадии р-ции. Напр., для нитрования соед. I (Y=Н) в ацетоне при 25 °С r=-7,3, для замещения Y=F в соед. I на метоксигруппу под действием CH3ONa r=+7,55, для этерификации бензойных к-т II метиловым спиртом r=-0,58. Рассчитанные по данным о константах диссоциации бензойных к-т II значения а для заместителей, способных к сопряжению с реакц. центрами Y в соединениях I, оказываются заниженными по абс. величине. Такие заместители, как NO2, CHO, СООСН3, взаимодействующие с сильными нуклеоф. группами Y=ОН, О-, СH2-, NH2 и др., или электронодонорные заместители ОН, О-, NH2, SR, взаимодействующие с электроф. реакц. центрами Y=COR, NO2, С+ и др., характеризуются соотв. нуклеоф. s - и электроф. s+ константами, вычисляемыми на основе подходящих реакц. серий. Применение констант s - и s+ позволяет сохранить форму К. с. (5). Более точные корреляции достигаются при переходе к многопараметровым ур-ниям, в к-рые включаются константы заместителей, характеризующие отдельно индуктивное, мезомерное и стерич. влияние заместителей. Если оставить в реакц. серии соед. II в качестве заместителей только группы, своб. от сопряжения с реакц. центром (ОСН3, F, Cl, Вr, I, СОСН3), можно получить ряд констант s°, значения к-рых учитывают индукц. эффект заместителя в бензольном кольце и индуцируемую поляризацию p-электронов кольца. Посредством подбора подходящих модельных соед. (напр., 4-замещенных бицикло [2,2,2] октан-1-карбоновых к-т) можно, пользуясь соотношением типа (5), получить константы, описывающие только индукц. влияние sI, и выделить мезомерные константы заместителей sс и s0c:
sс=sn-sI s0c=s0n-sI,
где s0n - константа заместителя в пара-положении. Корреляц. четырехпараметровые ур-ния:
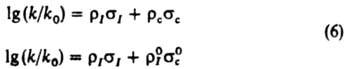
позволяют раздельно оценивать вклады индукц. и мезомерных эффектов заместителей в относит. реакц. способность. Известны другие К. с. [Юкава-Цуно, Дьюара - Грисдейла, Свена(Свейна) - Лаптона], в к-рых применяются др. подходы к разделению общего эффекта заместителя на его составляющие. Рассмотренные константы заместителей характеристичны для производных бензола I. Развиты К. с. для характеристики реакц. способности др. типов соед. - полиядерных ароматич., гетероциклич., олефинов и алифатич. соединений. В последнем случае применяют ур-ние Тафта [s* - константа Тафта, k и kCH3 - константы скорости или равновесия р-ций алифатич. соед. соотв. RY и CH3Y, r* - параметр, аналогичный r в ур-нии (4)]:

и для соед. с неск. заместителями:
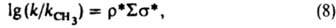
причем s* линейно связаны с константами sI:
s*(R)=6,23sI(R) sI(R)=0,45s*(CH2R)
Для учета влияния пространств. эффектов заместителей на реакц. способность алифатич. соед. вводятся стерич. константы заместителей Es, определяемые по данным о скоростях кислотного гидролиза сложных эфиров: Es=lg(k/k0), где k иk0 - константы скорости соотв. для. соед. с заместителем R в ацильной компоненте и для ацетата. Константы Es определяются объемом заместителей и линейно зависят от их ван-дер-ваальсовых радиусов. Совместный учет влияния электронных и пространств. факторов на реакц. способность алифатич. соед. осуществляется при помощи ур-ния:
lg(k/k0)=r*s*+rsEs, (9) где rs - параметр, аналогичный по смыслу r в ур-нии (4).
К. с. для вариации реагентов применяют для количеств, описания р-ций с варьируемым реагентом при постоянном субстрате. Для бимолекулярного нуклеоф. замещения выполняется ур-ние Свена-Скотта: lg(k/k0)=s.n. (10) где k и k0 - константы скорости р-ции субстрата соотв. с данным нуклеофилом и с водой, параметр s характеризует чувствительность р-ции к варьированию реагента (селективность), константы п определяются по данным о кинетике р-ций стандартного субстрата - СН3Вr с рядом нуклеофилов и зависят от условий проведения р-ции (среды, т-ры). Для водных р-ров при 0 °С значения п для нек-рых нуклеофилов равны 6,35 (для S2О32-, 5,13 (CN-), 4,16 (NH3), 2,99 (Сl-), 1,88 (F-), 0 (Н2O). Для р-ций стабильных орг. катионов и активир. карбонильных соед. с нуклеофилами хорошая корреляция достигается при помощи ур-ния Ритчи: lgk=N++C, (11) где С - константа, характерная для субстрата [напр., С=-5 для трис-(n-диметиламинофенил)метильного катиона и С - 1 для n-нитрофенилдиазония], значение N+ - ф-ции среды и т-ры. Так, N+=13,1 для нуклеофила C6H5S- в диметилсульфоксиде, а в СН3ОН составляет 10,7. Отсутствие в ур-нии (11) константы чувствительности р-ции означает, что два субстрата (1 и 2) имеют одинаковую селективность по отношению к одному и тому же нуклеофилу: lg(k1/k2)=С1-С2. Известен целый ряд многопараметровых К. с. для учета влияния варьируемого реагента на реакц. способность. К. с. для характеристики влияния р-рителя на реакц. способность. Соотношения ЛСЭ выполняются для характеристики влияния варьирования р-рителя на скорость или константы равновесия заданной р-ции. Для корреляции скоростей сольволиза используют ур-ние Грюнвальда - Уинстейна: lg(k/k0)=m.Y, (12) где т-константа чувствительности, аналогичная реакц. константе r в ур-ниях (4)-(9), а Y-параметр полярности р-рителя. В качестве стандартной серии (т =I) выбран сольволиз трет-С4Н9Сl в 80%-ном этаноле. Применимость ур-ния (12) ограничивается сольволизом в достаточно высокополярных р-рителях (Y=0 для 80%-ного этанола, -0,60 для формамида, -3,26 для трет-бутанола, 3,49 для воды). Для более широкого круга р-ций применяют многопараметровые К. с., включающие параметры р-рителей, рассчитанные на основе спектральных данных (смещения длинноволновых полос поглощения полярных соед. в разл. р-рителях), - параметры Димрота FT, константы Косовера Z, константы Тафта -Камле p*. В основе К. с. для физ. характеристик лежит установленная квантовомех. расчетами линейная зависимость констант заместителей а от величин электронных зарядов, индуцируемых заместителем на реакц. центре и прилегающих атомах, и др. характеристик электронного распределения в молекулах и ионах. Известны разнообразные К. с., связывающие дипольные моменты, частоты и интенсивности полос в колебат. спектрах, хим. сдвиги ядер в спектрах ЯМР и пр. от разл. типов электронных и стерич. констант заместителей. Напр., для хим. сдвигов 19F в спектрах ЯМР мета-(Fм) и пара-замещенных (Fn) бензолов с хорошей точностью выполняются К. с.:
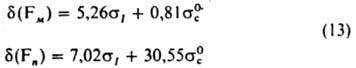
Для интенсивностей колебаний кольца производных бензола в области 1600 и 1585 см-1, характеризующих искажения p-электронной системы кольца, выполняется К. с.:
(А-100)1/2= 132,7sc°, (14)
где А - интегральная интенсивность полосы поглощения в ИК спектре. Соотношения (13), (14) используют для вычисления констант заместителей sI, s°c, s°. К. с. в биохимии становятся важным ср-вом предсказания и целенаправленного поиска структурных модификаций, способствующих повышению биол. активности, в сериях соед. с систематич. варьируемыми структурными признаками. Важный параметр, связываемый с библ. активностью, - коэф. распределения Р в бинарной системе углеводород-вода. Значения Р определены для более чем 10000 соединений. Общая модель К. с. типа "структура - активность" строится на изучении зависимостей (15)-(17). Скорость биол. ответа = B.C.k. (15) где С - кол-во введенного в организм препарата, В - фактор, характеризующий вероятность достижения препаратом рецептора за определенный промежуток времени, k - константа скорости или равновесия лимитирующей стадии р-ции, ведущей к контролируемому биол. ответу. Ур-ниe (15) сводится к К. с.: lg(1/C)=a(lgP)2+b(lgP)+clgk+d, (16) где a, b, с, d - коэф., получаемые в результате статистич. обработки данных эксперимента. Для мн. биол. систем для констант k выполняется К. с.:
lgk=ap+bv+cEs+d (17)
где p=lg(P/PH), Рн - коэф. распределения для соед. с заместителем Н. Ур-ния типа (15)-(17) - важные элементы системного анализа биол. активности орг. и прир. соединений. Лит.: Жданов Ю. А., Минкин В. И., Корреляционный анализ в органической химии. Ростов н/Д., 1966; Пальм В. А., Основы количественной теории органических реакций. Л., 1967; Джонсон К., Уравнения Гаммета, пер. с англ., М.. 1977; Advances in linear free energy relationships, ed. by N. B. Chapman. J. Shorter. N.Y.-L, 1972; Correlation analysis in chemistry. Recent advances, ed. by N.B. Chapman, J. Shorter, N. Y.-L, 1978; Hansch C, Leo A., Substituent constants for correlation analysis in chemistry and biology, N. Y., 1979; Klumpp G. W., Reactivity in organic chemistry, N. Y., 1982. В.И. Mинкин.
lgke=algKa+c, (1)
где а и с - эмпирич. параметры для данной р-ции. Ур-ние (1) - пример выполнения общего соотношения линейности своб. энергий (ЛСЭ), согласно к-рому изменения в своб. энергиях р-ций DG° и в своб. энергиях активации (DG++), вызываемые в разл. р-циях одинаковыми вариациями структуры реагирующих соед. или среды, связаны линейными зависимостями:
dRDGi=adRDGj+b, (2)
где dR - оператор изменения структуры (напр., при введении заместителя R) или среды, i, j - индексы р-ций. Ур-ние (2) отражает широкую эмпирич. закономерность: сходные изменения в строении соед. приводят к сходным изменениям в реакц. способности. Оно не вытекает из принципов термодинамики, поэтому К. с., основанные на соотношении ЛСЭ, принято относить к экстратермодинамическим. Важнейшее К. с. этого типа - ур-ние Гаммета, сопоставляющее изменения в константах скорости или равновесия, индуцируемые введением заместителей в ароматич. бензольное ядро в соед. I, с изменениями в константах диссоциации бензойных к-т II при аналогичном замещении:
dRDGi=аdRDG0БK, (3)
где DGi - своб. энергии р-ции или активации произвольной р-ции соед. I в произвольных (но одинаковых для данной серии-,соед.) условиях, а DG0БК - своб. энергия диссоциации бензойной к-ты в заданных (не обязательно идентичных) условиях. Ур-ние (3) сводится к виду:
lg(k/k0)=rlg(K/K0)БК (4)
где k - константа скорости (равновесия) р-ции соед. I с заместителем R, а k0 - та же константа для соед. с R=Н, К и К0 - константы диссоциации соответствующих бензойных к-т II, r - константа, характеризующая относительную (в сравнении с эталонной серией р-ций диссоциации бензойных к-т) чувствительность р-ций соед. I к структурным изменениям.
Реакц. серия диссоциации бензойных к-т в воде при 25 °С выбрана как стандартная, при этом величины lg(K/K0)=s представляют константы заместителей R. Ур-ние (4) известно как ур-ние Гаммета и обычно записывается в форме:
lgk=rs+lgk0 (5)
Ур-ние (5) позволяет с точностью b15% рассчитывать кинетич. и равновесные параметры мн. р-ций производных бензола с мета- и пaра-заместителями, располагая константами заместителей и величинами r, определенными статистически достоверно для неск. соед. данной реакц. серии. К. с. для структурных вариаций субстрата. В табл. приведены значения констант заместителей s для нек-рых наиб. важных групп (sм и sn - константы заместителей в мета- и пара-положениях). Для р-ций, ускоряемых электронодонорными заместителями (s<0), r<0, для р-ций, ускоряемых электроноакцепторными заместителями
(s>0), r>0. Абс. величина r тем больше, чем более полярно переходное состояние лимитирующей стадии р-ции. Напр., для нитрования соед. I (Y=Н) в ацетоне при 25 °С r=-7,3, для замещения Y=F в соед. I на метоксигруппу под действием CH3ONa r=+7,55, для этерификации бензойных к-т II метиловым спиртом r=-0,58. Рассчитанные по данным о константах диссоциации бензойных к-т II значения а для заместителей, способных к сопряжению с реакц. центрами Y в соединениях I, оказываются заниженными по абс. величине. Такие заместители, как NO2, CHO, СООСН3, взаимодействующие с сильными нуклеоф. группами Y=ОН, О-, СH2-, NH2 и др., или электронодонорные заместители ОН, О-, NH2, SR, взаимодействующие с электроф. реакц. центрами Y=COR, NO2, С+ и др., характеризуются соотв. нуклеоф. s - и электроф. s+ константами, вычисляемыми на основе подходящих реакц. серий. Применение констант s - и s+ позволяет сохранить форму К. с. (5). Более точные корреляции достигаются при переходе к многопараметровым ур-ниям, в к-рые включаются константы заместителей, характеризующие отдельно индуктивное, мезомерное и стерич. влияние заместителей. Если оставить в реакц. серии соед. II в качестве заместителей только группы, своб. от сопряжения с реакц. центром (ОСН3, F, Cl, Вr, I, СОСН3), можно получить ряд констант s°, значения к-рых учитывают индукц. эффект заместителя в бензольном кольце и индуцируемую поляризацию p-электронов кольца. Посредством подбора подходящих модельных соед. (напр., 4-замещенных бицикло [2,2,2] октан-1-карбоновых к-т) можно, пользуясь соотношением типа (5), получить константы, описывающие только индукц. влияние sI, и выделить мезомерные константы заместителей sс и s0c:
sс=sn-sI s0c=s0n-sI,
где s0n - константа заместителя в пара-положении. Корреляц. четырехпараметровые ур-ния:
позволяют раздельно оценивать вклады индукц. и мезомерных эффектов заместителей в относит. реакц. способность. Известны другие К. с. [Юкава-Цуно, Дьюара - Грисдейла, Свена(Свейна) - Лаптона], в к-рых применяются др. подходы к разделению общего эффекта заместителя на его составляющие. Рассмотренные константы заместителей характеристичны для производных бензола I. Развиты К. с. для характеристики реакц. способности др. типов соед. - полиядерных ароматич., гетероциклич., олефинов и алифатич. соединений. В последнем случае применяют ур-ние Тафта [s* - константа Тафта, k и kCH3 - константы скорости или равновесия р-ций алифатич. соед. соотв. RY и CH3Y, r* - параметр, аналогичный r в ур-нии (4)]:
и для соед. с неск. заместителями:
причем s* линейно связаны с константами sI:
s*(R)=6,23sI(R) sI(R)=0,45s*(CH2R)
Для учета влияния пространств. эффектов заместителей на реакц. способность алифатич. соед. вводятся стерич. константы заместителей Es, определяемые по данным о скоростях кислотного гидролиза сложных эфиров: Es=lg(k/k0), где k иk0 - константы скорости соотв. для. соед. с заместителем R в ацильной компоненте и для ацетата. Константы Es определяются объемом заместителей и линейно зависят от их ван-дер-ваальсовых радиусов. Совместный учет влияния электронных и пространств. факторов на реакц. способность алифатич. соед. осуществляется при помощи ур-ния:
lg(k/k0)=r*s*+rsEs, (9) где rs - параметр, аналогичный по смыслу r в ур-нии (4).
К. с. для вариации реагентов применяют для количеств, описания р-ций с варьируемым реагентом при постоянном субстрате. Для бимолекулярного нуклеоф. замещения выполняется ур-ние Свена-Скотта: lg(k/k0)=s.n. (10) где k и k0 - константы скорости р-ции субстрата соотв. с данным нуклеофилом и с водой, параметр s характеризует чувствительность р-ции к варьированию реагента (селективность), константы п определяются по данным о кинетике р-ций стандартного субстрата - СН3Вr с рядом нуклеофилов и зависят от условий проведения р-ции (среды, т-ры). Для водных р-ров при 0 °С значения п для нек-рых нуклеофилов равны 6,35 (для S2О32-, 5,13 (CN-), 4,16 (NH3), 2,99 (Сl-), 1,88 (F-), 0 (Н2O). Для р-ций стабильных орг. катионов и активир. карбонильных соед. с нуклеофилами хорошая корреляция достигается при помощи ур-ния Ритчи: lgk=N++C, (11) где С - константа, характерная для субстрата [напр., С=-5 для трис-(n-диметиламинофенил)метильного катиона и С - 1 для n-нитрофенилдиазония], значение N+ - ф-ции среды и т-ры. Так, N+=13,1 для нуклеофила C6H5S- в диметилсульфоксиде, а в СН3ОН составляет 10,7. Отсутствие в ур-нии (11) константы чувствительности р-ции означает, что два субстрата (1 и 2) имеют одинаковую селективность по отношению к одному и тому же нуклеофилу: lg(k1/k2)=С1-С2. Известен целый ряд многопараметровых К. с. для учета влияния варьируемого реагента на реакц. способность. К. с. для характеристики влияния р-рителя на реакц. способность. Соотношения ЛСЭ выполняются для характеристики влияния варьирования р-рителя на скорость или константы равновесия заданной р-ции. Для корреляции скоростей сольволиза используют ур-ние Грюнвальда - Уинстейна: lg(k/k0)=m.Y, (12) где т-константа чувствительности, аналогичная реакц. константе r в ур-ниях (4)-(9), а Y-параметр полярности р-рителя. В качестве стандартной серии (т =I) выбран сольволиз трет-С4Н9Сl в 80%-ном этаноле. Применимость ур-ния (12) ограничивается сольволизом в достаточно высокополярных р-рителях (Y=0 для 80%-ного этанола, -0,60 для формамида, -3,26 для трет-бутанола, 3,49 для воды). Для более широкого круга р-ций применяют многопараметровые К. с., включающие параметры р-рителей, рассчитанные на основе спектральных данных (смещения длинноволновых полос поглощения полярных соед. в разл. р-рителях), - параметры Димрота FT, константы Косовера Z, константы Тафта -Камле p*. В основе К. с. для физ. характеристик лежит установленная квантовомех. расчетами линейная зависимость констант заместителей а от величин электронных зарядов, индуцируемых заместителем на реакц. центре и прилегающих атомах, и др. характеристик электронного распределения в молекулах и ионах. Известны разнообразные К. с., связывающие дипольные моменты, частоты и интенсивности полос в колебат. спектрах, хим. сдвиги ядер в спектрах ЯМР и пр. от разл. типов электронных и стерич. констант заместителей. Напр., для хим. сдвигов 19F в спектрах ЯМР мета-(Fм) и пара-замещенных (Fn) бензолов с хорошей точностью выполняются К. с.:
Для интенсивностей колебаний кольца производных бензола в области 1600 и 1585 см-1, характеризующих искажения p-электронной системы кольца, выполняется К. с.:
(А-100)1/2= 132,7sc°, (14)
где А - интегральная интенсивность полосы поглощения в ИК спектре. Соотношения (13), (14) используют для вычисления констант заместителей sI, s°c, s°. К. с. в биохимии становятся важным ср-вом предсказания и целенаправленного поиска структурных модификаций, способствующих повышению биол. активности, в сериях соед. с систематич. варьируемыми структурными признаками. Важный параметр, связываемый с библ. активностью, - коэф. распределения Р в бинарной системе углеводород-вода. Значения Р определены для более чем 10000 соединений. Общая модель К. с. типа "структура - активность" строится на изучении зависимостей (15)-(17). Скорость биол. ответа = B.C.k. (15) где С - кол-во введенного в организм препарата, В - фактор, характеризующий вероятность достижения препаратом рецептора за определенный промежуток времени, k - константа скорости или равновесия лимитирующей стадии р-ции, ведущей к контролируемому биол. ответу. Ур-ниe (15) сводится к К. с.: lg(1/C)=a(lgP)2+b(lgP)+clgk+d, (16) где a, b, с, d - коэф., получаемые в результате статистич. обработки данных эксперимента. Для мн. биол. систем для констант k выполняется К. с.:
lgk=ap+bv+cEs+d (17)
где p=lg(P/PH), Рн - коэф. распределения для соед. с заместителем Н. Ур-ния типа (15)-(17) - важные элементы системного анализа биол. активности орг. и прир. соединений. Лит.: Жданов Ю. А., Минкин В. И., Корреляционный анализ в органической химии. Ростов н/Д., 1966; Пальм В. А., Основы количественной теории органических реакций. Л., 1967; Джонсон К., Уравнения Гаммета, пер. с англ., М.. 1977; Advances in linear free energy relationships, ed. by N. B. Chapman. J. Shorter. N.Y.-L, 1972; Correlation analysis in chemistry. Recent advances, ed. by N.B. Chapman, J. Shorter, N. Y.-L, 1978; Hansch C, Leo A., Substituent constants for correlation analysis in chemistry and biology, N. Y., 1979; Klumpp G. W., Reactivity in organic chemistry, N. Y., 1982. В.И. Mинкин.