


КИНЕТИКА ХИМИЧЕСКАЯ (от греч. kinetikos-движущий), раздел физ. химии, изучающий хим. р-цию как процесс, протекающий во времени, механизм этого процесса, его зависимость от условий осуществления. К. х. устанавливает временные закономерности протекания хим. р-ций, связь между скоростью р-ции и условиями ее проведения, выявляет факторы, влияющие на скорость и направление хим. р-ций. Изучить механизм сложного хим. процесса - означает выяснить, из каких элементарных стадий он состоит и каким образом элементарные стадии связаны друг с другом, какие образуются промежут. продукты и т. п. Теорегич. К. х. занимается построением мат. моделей сложных хим. процессов, анализом этих моделей в сопоставлении с эксперим. данными. Важной задачей К. х. является изучение элементарных р-ций с участием активных частиц: своб. атомов и радикалов, ионов и ион-радикалов, возбужденных молекул и др. Используя результаты кинетич. исследований и изучения строения молекул и хим. связи, К. х. устанавливает связь между строением молекул реагентов и их реакц. способностью. Динамика элементарного акта изучает теоретич. и эксперимент. методами элементарный акт хим. р-ции и предшествующие ему механизмы возбуждения реагирующих частиц. Кинетич. исследования входят как важная составная часть во многие самостоят, разделы химии, такие, как катализ, фотохимия, плазмохимия, радиационная химия, электрохимия и др. В своих методах исследования и теоретич. обобщениях К. х. использует достижения математики, кибернетики, атомной и мол. физики, квантовой химии, спектроскопии, аналит. химии. Кинетич. данные и теоретич. концепции К. х. используются при создании экологич. моделей атмосферы и гидросферы, при анализе процессов, происходящих в космосе.
Основные понятия К. х. Любая хим. р-ция представляет собой совокупность элементарных актов хим. превращения. Каждый такой акт есть превращение одной или неск. находящихся в контакте или взаимодействии частиц реагентов в частицы продуктов. Простые р-ции состоят из однотипных элементарных актов. В зависимости от числа частиц, принимающих участие в р-ции, они делятся на мономолекулярные реакции, бимолекулярные реакции и тримолекулярные реакции. Р-ции, при протекании к-рых осуществляются разнотипные элементарные акты, наз. сложными реакииями. К ним относятся обратимые, параллельные, последовательные и др. многостадийные р-ции, цепные реакции, сопряженные реакции и др. За развитием хим. р-ций следят по изменению концентраций реагирующих в-в и (или) продуктов, опытные данные представляют графически в виде кинетич. кривых концентрация время t (см. Кинетическое уравнение). Путем дифференцирования кинетич. кривой для реагента А получают скорость изменения его концентрации vА=-d[A]/dt. Скорость р-ции, согласно действующих масс закону, прямо пропорциональна произведению концентраций
ций участвующих в р-ции хим. соед. Для р-ции vAA+vBB:vYY+vzZ, где А и В - реагенты, Y и Z - продукты, vA, vB, 1 d[A] vY и vz - стехиометрия, коэф., скорость v=-(1/vA)(d[A]/dt)= k[А]nА [В]nB, где k - константа скорости р-ции, nА и nв- порядки реакции по реагентам А и В соотв.; суммарный порядок р-ции п=nА+nв. Для простой р-ции, как правило, nА=vA, nB=vB для сложной р-ции порядок и стехиометрич. коэф. могут и не совпадать.
Для характеристики протекания р-ции во времени используют, кроме скорости и константы скорости, такие параметры, как характеристич. время превращения (время, за к-рое концентрация реагента уменьшается в е раз), период полупревращения (время, в течение к-рого концентрация реагента уменьшается наполовину) и др. Автокаталитические (см. Автокатализ) и цепные р-ции часто имеют период индукции - отрезок времени, в течение к-рого не наблюдается заметного протекания р-ции. Зависимость константы скорости от т-ры Т обычно выражается в аррениусовской форме: k=Ae-E/RT, где А - предэкспоненц. множитель, Е - энергия активации р-ции, R - газовая постоянная (см. Аррениуса уравнение). Для бимолекулярных р-ций, согласно столкновений теории, А=z0P, где z0 - фактор частоты столкновений, Р - стерический фактор. Согласно активированного комплекса теории, 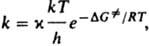 где ( - трансмиссионный коэф., k - постоянная Больцмана, h - постоянная Планка,
где ( - трансмиссионный коэф., k - постоянная Больцмана, h - постоянная Планка, 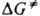 - энергия Гиббса активации р-ции (
- энергия Гиббса активации р-ции (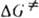 =
=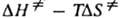 , где
, где 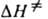 и
и 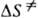 -энтальпия и энтропия активации р-ции). Скорость протеканий бимолекулярной р-ции в газовой фазе часто характеризуется сечением захвата s.
Методы К. х. Для изучения кинетики хим. р-ций широко используются разнообразные методы хим. анализа продуктов и реагентов, физ. методы контроля таких характеристик реагирующей системы, как объем, т-ра, плотность, спектроскопич., масс-спектромстрич., элсктрохим., хроматографич. методы. Часто в опытах изменяют концентрации реагентов, т-ру, давление, магн. поле, вязкость среды, площадь пов-сти реакц. сосуда. В систему, где протекает р-ция, вводят как в начале опыта, так и по ходу опыта инициаторы радикальные, ингибиторы, катализаторы, промежут. или конечные продукты. Для изучения превращения отдельных фрагментов молекулы используют реагенты с изотопными метками, оптически активные реагенты, воздействуют на систему лазерным излучением. При изучении цепных и нецепных радикальных р-ций используют акцепторы своб. радикалов и вещества-ловушки своб. радикалов (см. Спиновых ловушек метод). Р-ции активных (быстро превращающихся) частиц изучают спец. кинетич. методами (см. Адиабатического сжатия метод, Диффузионных пламен метод, Конкурирующих реакций метод, Молекулярных пучков метод, Релаксационные методы, Струевые кинетические методы, Ударных труб метод).
Теоретич. анализ эксперим. данных проводят с использованием методов совр. вычислит. математики. Мат. методы используются для решения т. наз. прямой задачи К. х. расчета кинетич. поведения продуктов и реагентов при заданной схеме р-ции и известной константе скорости и обратной задачи К. х. воссоздания схемы р-ции и кинетич. параметров по эксперим. данным.
Реакции в газе, жидкости, твердом теле. Изучение газофазных р-ций имеет важное значение для теоретич. К. х., так как в газе можно наблюдать р-цию в "чистом виде" и исключить (или выявить) роль частиц, не вступающих в р-цию. Большой эксперим. материал накоплен по мономолекулярным р-циям; для них разработана статистич. теория Раиса Рамспергера Касссля-Маркуса, к-рая позволяет рассчитать константу скорости распада в условиях максвелл-больцмановского равновесия. Много кинетич. данных в виде констант скорости получено для бимолекулярных р-ций с участием малоатомных молекул,
радикалов и ионов. Р-ции рекомбинации и присоединения атомов и радикалов осуществляются при условии стабилизации возбужденной молекулы продукта в результате столкновения с третьей частицей. Теоретич. расчет константы скорости бимолекулярной р-ции - сложная задача, к-рая включает расчет поверхности потенциальной энергии и движения ядер частиц-реагентов по этой пов-сти. Для решения этой задачи разработаны неэмпирич. и полуэмпирич. методы. Важное место занимает изучение зависимости скорости или константы скорости газофазных моно- и бимолекулярных р-ций от давления, т. к. от частоты столкновений частиц зависит их активация.
Р-ции, протекающие в жидкой фазе, чрезвычайно разнообразны как по строению реагентов, так и по механизмам превращения (см. Реакции в жидкостях). При диссоциации молекулы на свобод. радикалы и атомы наблюдается клетки эффект. Медленная (в сравнении с газом) диффузия частиц в жидкости приводит к тому, что безактивационные бимолекулярные р-ции протекают как диффузионно-контролируемые реакции. Р-ции, имеющие значит. энергию активации, протекают, как правило, в кинетич. режиме. Реагенты в р-ре часто образуют между собой мол. комплексы и разнообразные ассоциаты. Это отражается на кинетике р-ции и часто существенно меняет кинетич. закономерности процесса. Полярный р-ритель облегчает ионизацию молекулы, в р-ре появляются контактные и разделенные ионные пары. Возникает вероятность параллельного протекания р-ции по разным механизмам. Нередко, однако, р-ция протекает по мол. механизму как самосогласованный процесс перестройки мол. орбиталей реагирующих частиц (см. Вудворда Хофмана правила). Окислит.-восстановит. р-ции могут происходить в жидкости по механизму квантового туннелирования (см. Туннельный эффект).
В хим. технологии широко распространены гетерофазные р-ции, при к-рых реагенты находятся в разных фазах, а р-ция протекает на границе раздела фаз. Скорость таких р-ций зависит от площади пов-сти раздела фаз, коэф. диффузии реагентов и продуктов, условий перемешивания, кинетич. параметров р-ции. Теоретич. описание кинетики таких р-ций связано с решением диффузионной задачи (см. Макрокинетика).
Хим. р-ции в твердой фазе отличаются тем, что продукты образуют новую фазу и протекание р-ции осложнено зарождением новой фазы. Р-ция протекает на границе раздела фаз. Ее обычно характеризуют степенью превращения x, а скорость р-ции - производной dx/dt. Кинетич. кривая имеет S-образный характер. Важную роль в возникновении новой фазы играют дефекты кристаллич. решетки (см. Реакции в твердых телах).
Среди сложных р-ции широко распространены цепные р-ции, при к-рых образующийся в системе активный центр (своб. атом, радикал, ион, ион-радикал) вызывает циклически повторяющуюся цепочку превращений реагентов в продукты. В сложном цепном процессе выделяют стадии зарождения, продолжения, разветвления и обрыва цепи. Различают р-ции неразветвленные, разветвленные, с энер-гетич. разветвлением и вырожденно-разветвленные. По цепному механизму протекает распад мн. молекул, в т.ч. крекинг углеводородов, окисление орг. соед. молекулярным кислoродом, радикальная и ионная полимеризации, хлорирование, бромирование, сульфохлорирование и т.п.
-энтальпия и энтропия активации р-ции). Скорость протеканий бимолекулярной р-ции в газовой фазе часто характеризуется сечением захвата s.
Методы К. х. Для изучения кинетики хим. р-ций широко используются разнообразные методы хим. анализа продуктов и реагентов, физ. методы контроля таких характеристик реагирующей системы, как объем, т-ра, плотность, спектроскопич., масс-спектромстрич., элсктрохим., хроматографич. методы. Часто в опытах изменяют концентрации реагентов, т-ру, давление, магн. поле, вязкость среды, площадь пов-сти реакц. сосуда. В систему, где протекает р-ция, вводят как в начале опыта, так и по ходу опыта инициаторы радикальные, ингибиторы, катализаторы, промежут. или конечные продукты. Для изучения превращения отдельных фрагментов молекулы используют реагенты с изотопными метками, оптически активные реагенты, воздействуют на систему лазерным излучением. При изучении цепных и нецепных радикальных р-ций используют акцепторы своб. радикалов и вещества-ловушки своб. радикалов (см. Спиновых ловушек метод). Р-ции активных (быстро превращающихся) частиц изучают спец. кинетич. методами (см. Адиабатического сжатия метод, Диффузионных пламен метод, Конкурирующих реакций метод, Молекулярных пучков метод, Релаксационные методы, Струевые кинетические методы, Ударных труб метод).
Теоретич. анализ эксперим. данных проводят с использованием методов совр. вычислит. математики. Мат. методы используются для решения т. наз. прямой задачи К. х. расчета кинетич. поведения продуктов и реагентов при заданной схеме р-ции и известной константе скорости и обратной задачи К. х. воссоздания схемы р-ции и кинетич. параметров по эксперим. данным.
Реакции в газе, жидкости, твердом теле. Изучение газофазных р-ций имеет важное значение для теоретич. К. х., так как в газе можно наблюдать р-цию в "чистом виде" и исключить (или выявить) роль частиц, не вступающих в р-цию. Большой эксперим. материал накоплен по мономолекулярным р-циям; для них разработана статистич. теория Раиса Рамспергера Касссля-Маркуса, к-рая позволяет рассчитать константу скорости распада в условиях максвелл-больцмановского равновесия. Много кинетич. данных в виде констант скорости получено для бимолекулярных р-ций с участием малоатомных молекул,
радикалов и ионов. Р-ции рекомбинации и присоединения атомов и радикалов осуществляются при условии стабилизации возбужденной молекулы продукта в результате столкновения с третьей частицей. Теоретич. расчет константы скорости бимолекулярной р-ции - сложная задача, к-рая включает расчет поверхности потенциальной энергии и движения ядер частиц-реагентов по этой пов-сти. Для решения этой задачи разработаны неэмпирич. и полуэмпирич. методы. Важное место занимает изучение зависимости скорости или константы скорости газофазных моно- и бимолекулярных р-ций от давления, т. к. от частоты столкновений частиц зависит их активация.
Р-ции, протекающие в жидкой фазе, чрезвычайно разнообразны как по строению реагентов, так и по механизмам превращения (см. Реакции в жидкостях). При диссоциации молекулы на свобод. радикалы и атомы наблюдается клетки эффект. Медленная (в сравнении с газом) диффузия частиц в жидкости приводит к тому, что безактивационные бимолекулярные р-ции протекают как диффузионно-контролируемые реакции. Р-ции, имеющие значит. энергию активации, протекают, как правило, в кинетич. режиме. Реагенты в р-ре часто образуют между собой мол. комплексы и разнообразные ассоциаты. Это отражается на кинетике р-ции и часто существенно меняет кинетич. закономерности процесса. Полярный р-ритель облегчает ионизацию молекулы, в р-ре появляются контактные и разделенные ионные пары. Возникает вероятность параллельного протекания р-ции по разным механизмам. Нередко, однако, р-ция протекает по мол. механизму как самосогласованный процесс перестройки мол. орбиталей реагирующих частиц (см. Вудворда Хофмана правила). Окислит.-восстановит. р-ции могут происходить в жидкости по механизму квантового туннелирования (см. Туннельный эффект).
В хим. технологии широко распространены гетерофазные р-ции, при к-рых реагенты находятся в разных фазах, а р-ция протекает на границе раздела фаз. Скорость таких р-ций зависит от площади пов-сти раздела фаз, коэф. диффузии реагентов и продуктов, условий перемешивания, кинетич. параметров р-ции. Теоретич. описание кинетики таких р-ций связано с решением диффузионной задачи (см. Макрокинетика).
Хим. р-ции в твердой фазе отличаются тем, что продукты образуют новую фазу и протекание р-ции осложнено зарождением новой фазы. Р-ция протекает на границе раздела фаз. Ее обычно характеризуют степенью превращения x, а скорость р-ции - производной dx/dt. Кинетич. кривая имеет S-образный характер. Важную роль в возникновении новой фазы играют дефекты кристаллич. решетки (см. Реакции в твердых телах).
Среди сложных р-ции широко распространены цепные р-ции, при к-рых образующийся в системе активный центр (своб. атом, радикал, ион, ион-радикал) вызывает циклически повторяющуюся цепочку превращений реагентов в продукты. В сложном цепном процессе выделяют стадии зарождения, продолжения, разветвления и обрыва цепи. Различают р-ции неразветвленные, разветвленные, с энер-гетич. разветвлением и вырожденно-разветвленные. По цепному механизму протекает распад мн. молекул, в т.ч. крекинг углеводородов, окисление орг. соед. молекулярным кислoродом, радикальная и ионная полимеризации, хлорирование, бромирование, сульфохлорирование и т.п.
Исторический очерк. Первые кинетич. исследования с измерением скорости хим. р-ции выполнили Л. Вильгельми (гидролиз тростникового сахара, 1850), М. Бертло и Л. Пеан де Сен-Жиль (этерификация, 1862), Ф. Гаркур и В. Эссон (окисление щавелевой к-ты КМnО4, 1866). К. Гульдберг и П. Вааге сформулировали закон действующих масс (1862 1867); С Аррениус (1889) обосновал роль активных молекул в хим р-ции и сформулировал закон температурной зависимости константы скорости простых р-ций. Я Вант-Гофф получил ф-лы, описывающие протекание моно-, би- и три-молекулярных р-ций и обобщил эксперим. данные в первой монографии по К х (1884). Н. А. Меншуткин в 70-х гг 19 в. выполнил серию исследований по влиянию среды и строения молекул реагентов на скорость терификации и ввел в отечеств, научную лит. термин "химическая кинетика" (1888). В 30-х гг. 20 в. Г. Эйрингом и М. Поляни разработана теория активир. комплекса. Представления о важной роли промежут. продуктов в протекании сложных р-ций вошла в К. х. одновременно . с перекисной теорией окисления Баха -Энглера (1897) и теорией сопряженных р-ций окисления Лютера - Шилова (1903). Цепные неразветвленные р-ции были открыты М. Боденштейном (1913). X. Бекстрём показал, что ингибиторы тормозят цепные р-ции, обрывая цепи (1926). Цепные разветвленные р-ции были открыты Н. Н. Семеновым и С. Хиншелвудом (1926-28). Важный вклад в развитие кинетики цепных р-ций внесли В. Н. Кондратьев, В. В. Воеводский, А. Б. Налбандян, Н. М. Эмануэль. Для современной К. х. характерно широкое использование разнообразных методов изучения быстропротекающих хим. процессов, автоматизация эксперимента, использование ЭВМ для обработки эксперим. данных. Кинетич. информация собирается, хранится и используется через банки кинетич. констант. Интенсивно развивается динамика элементарного акта как теоретич. направление К. х. и (с применением ЭВМ) новый эксперим. метод. В неравновесной химической кинетике изучаются процессы передачи энергии и активации молекул. Важное значение приобрели лазерные методы для возбуждения молекул и для контроля за протеканием р-ции (см. Лазерная химия). Возрос интерес к изучению кинетики р-ций в экстремальных условиях, напр. при мех. разрушении в-ва, низких т-рах (см. Механохимия, Криохимия). Лит.. Вант-Гофф Я. Г., Очерки по химической динамике, в его кн.. Избранные труды по химии, М., 1984; Семенов Н. Н., Цепные реакции, 2 изд., М., 1986; Мелвин-Хьюз Э. А., Равновесие и кинетика реакций в растворах, М., 1975; Кондратьев В.Н., Никитин Е.Е.. Химические процессы в газах, М., 1981; Эйринг Г., Лин С. Г., Лин С М., Основы химической кинетики, пер. с англ., М., 1983; Эмануэль Н. М., Кнорре Д. Г, Курс химической кинетики, М., 1985; Денисов Е.Т., Кинетика гомогенных химических реакций, М., 1988. Е. Т Денисов.
где ( - трансмиссионный коэф., k - постоянная Больцмана, h - постоянная Планка, - энергия Гиббса активации р-ции (=, где и -энтальпия и энтропия активации р-ции). Скорость протеканий бимолекулярной р-ции в газовой фазе часто характеризуется сечением захвата s.
Методы К. х. Для изучения кинетики хим. р-ций широко используются разнообразные методы хим. анализа продуктов и реагентов, физ. методы контроля таких характеристик реагирующей системы, как объем, т-ра, плотность, спектроскопич., масс-спектромстрич., элсктрохим., хроматографич. методы. Часто в опытах изменяют концентрации реагентов, т-ру, давление, магн. поле, вязкость среды, площадь пов-сти реакц. сосуда. В систему, где протекает р-ция, вводят как в начале опыта, так и по ходу опыта инициаторы радикальные, ингибиторы, катализаторы, промежут. или конечные продукты. Для изучения превращения отдельных фрагментов молекулы используют реагенты с изотопными метками, оптически активные реагенты, воздействуют на систему лазерным излучением. При изучении цепных и нецепных радикальных р-ций используют акцепторы своб. радикалов и вещества-ловушки своб. радикалов (см. Спиновых ловушек метод). Р-ции активных (быстро превращающихся) частиц изучают спец. кинетич. методами (см. Адиабатического сжатия метод, Диффузионных пламен метод, Конкурирующих реакций метод, Молекулярных пучков метод, Релаксационные методы, Струевые кинетические методы, Ударных труб метод).
Теоретич. анализ эксперим. данных проводят с использованием методов совр. вычислит. математики. Мат. методы используются для решения т. наз. прямой задачи К. х. расчета кинетич. поведения продуктов и реагентов при заданной схеме р-ции и известной константе скорости и обратной задачи К. х. воссоздания схемы р-ции и кинетич. параметров по эксперим. данным.
Реакции в газе, жидкости, твердом теле. Изучение газофазных р-ций имеет важное значение для теоретич. К. х., так как в газе можно наблюдать р-цию в "чистом виде" и исключить (или выявить) роль частиц, не вступающих в р-цию. Большой эксперим. материал накоплен по мономолекулярным р-циям; для них разработана статистич. теория Раиса Рамспергера Касссля-Маркуса, к-рая позволяет рассчитать константу скорости распада в условиях максвелл-больцмановского равновесия. Много кинетич. данных в виде констант скорости получено для бимолекулярных р-ций с участием малоатомных молекул,
радикалов и ионов. Р-ции рекомбинации и присоединения атомов и радикалов осуществляются при условии стабилизации возбужденной молекулы продукта в результате столкновения с третьей частицей. Теоретич. расчет константы скорости бимолекулярной р-ции - сложная задача, к-рая включает расчет поверхности потенциальной энергии и движения ядер частиц-реагентов по этой пов-сти. Для решения этой задачи разработаны неэмпирич. и полуэмпирич. методы. Важное место занимает изучение зависимости скорости или константы скорости газофазных моно- и бимолекулярных р-ций от давления, т. к. от частоты столкновений частиц зависит их активация.
Р-ции, протекающие в жидкой фазе, чрезвычайно разнообразны как по строению реагентов, так и по механизмам превращения (см. Реакции в жидкостях). При диссоциации молекулы на свобод. радикалы и атомы наблюдается клетки эффект. Медленная (в сравнении с газом) диффузия частиц в жидкости приводит к тому, что безактивационные бимолекулярные р-ции протекают как диффузионно-контролируемые реакции. Р-ции, имеющие значит. энергию активации, протекают, как правило, в кинетич. режиме. Реагенты в р-ре часто образуют между собой мол. комплексы и разнообразные ассоциаты. Это отражается на кинетике р-ции и часто существенно меняет кинетич. закономерности процесса. Полярный р-ритель облегчает ионизацию молекулы, в р-ре появляются контактные и разделенные ионные пары. Возникает вероятность параллельного протекания р-ции по разным механизмам. Нередко, однако, р-ция протекает по мол. механизму как самосогласованный процесс перестройки мол. орбиталей реагирующих частиц (см. Вудворда Хофмана правила). Окислит.-восстановит. р-ции могут происходить в жидкости по механизму квантового туннелирования (см. Туннельный эффект).
В хим. технологии широко распространены гетерофазные р-ции, при к-рых реагенты находятся в разных фазах, а р-ция протекает на границе раздела фаз. Скорость таких р-ций зависит от площади пов-сти раздела фаз, коэф. диффузии реагентов и продуктов, условий перемешивания, кинетич. параметров р-ции. Теоретич. описание кинетики таких р-ций связано с решением диффузионной задачи (см. Макрокинетика).
Хим. р-ции в твердой фазе отличаются тем, что продукты образуют новую фазу и протекание р-ции осложнено зарождением новой фазы. Р-ция протекает на границе раздела фаз. Ее обычно характеризуют степенью превращения x, а скорость р-ции - производной dx/dt. Кинетич. кривая имеет S-образный характер. Важную роль в возникновении новой фазы играют дефекты кристаллич. решетки (см. Реакции в твердых телах).
Среди сложных р-ции широко распространены цепные р-ции, при к-рых образующийся в системе активный центр (своб. атом, радикал, ион, ион-радикал) вызывает циклически повторяющуюся цепочку превращений реагентов в продукты. В сложном цепном процессе выделяют стадии зарождения, продолжения, разветвления и обрыва цепи. Различают р-ции неразветвленные, разветвленные, с энер-гетич. разветвлением и вырожденно-разветвленные. По цепному механизму протекает распад мн. молекул, в т.ч. крекинг углеводородов, окисление орг. соед. молекулярным кислoродом, радикальная и ионная полимеризации, хлорирование, бромирование, сульфохлорирование и т.п.
Исторический очерк. Первые кинетич. исследования с измерением скорости хим. р-ции выполнили Л. Вильгельми (гидролиз тростникового сахара, 1850), М. Бертло и Л. Пеан де Сен-Жиль (этерификация, 1862), Ф. Гаркур и В. Эссон (окисление щавелевой к-ты КМnО4, 1866). К. Гульдберг и П. Вааге сформулировали закон действующих масс (1862 1867); С Аррениус (1889) обосновал роль активных молекул в хим р-ции и сформулировал закон температурной зависимости константы скорости простых р-ций. Я Вант-Гофф получил ф-лы, описывающие протекание моно-, би- и три-молекулярных р-ций и обобщил эксперим. данные в первой монографии по К х (1884). Н. А. Меншуткин в 70-х гг 19 в. выполнил серию исследований по влиянию среды и строения молекул реагентов на скорость терификации и ввел в отечеств, научную лит. термин "химическая кинетика" (1888). В 30-х гг. 20 в. Г. Эйрингом и М. Поляни разработана теория активир. комплекса. Представления о важной роли промежут. продуктов в протекании сложных р-ций вошла в К. х. одновременно . с перекисной теорией окисления Баха -Энглера (1897) и теорией сопряженных р-ций окисления Лютера - Шилова (1903). Цепные неразветвленные р-ции были открыты М. Боденштейном (1913). X. Бекстрём показал, что ингибиторы тормозят цепные р-ции, обрывая цепи (1926). Цепные разветвленные р-ции были открыты Н. Н. Семеновым и С. Хиншелвудом (1926-28). Важный вклад в развитие кинетики цепных р-ций внесли В. Н. Кондратьев, В. В. Воеводский, А. Б. Налбандян, Н. М. Эмануэль. Для современной К. х. характерно широкое использование разнообразных методов изучения быстропротекающих хим. процессов, автоматизация эксперимента, использование ЭВМ для обработки эксперим. данных. Кинетич. информация собирается, хранится и используется через банки кинетич. констант. Интенсивно развивается динамика элементарного акта как теоретич. направление К. х. и (с применением ЭВМ) новый эксперим. метод. В неравновесной химической кинетике изучаются процессы передачи энергии и активации молекул. Важное значение приобрели лазерные методы для возбуждения молекул и для контроля за протеканием р-ции (см. Лазерная химия). Возрос интерес к изучению кинетики р-ций в экстремальных условиях, напр. при мех. разрушении в-ва, низких т-рах (см. Механохимия, Криохимия). Лит.. Вант-Гофф Я. Г., Очерки по химической динамике, в его кн.. Избранные труды по химии, М., 1984; Семенов Н. Н., Цепные реакции, 2 изд., М., 1986; Мелвин-Хьюз Э. А., Равновесие и кинетика реакций в растворах, М., 1975; Кондратьев В.Н., Никитин Е.Е.. Химические процессы в газах, М., 1981; Эйринг Г., Лин С. Г., Лин С М., Основы химической кинетики,