


Особый класс циклич. ненасыщ. дикетонов - хиноны.
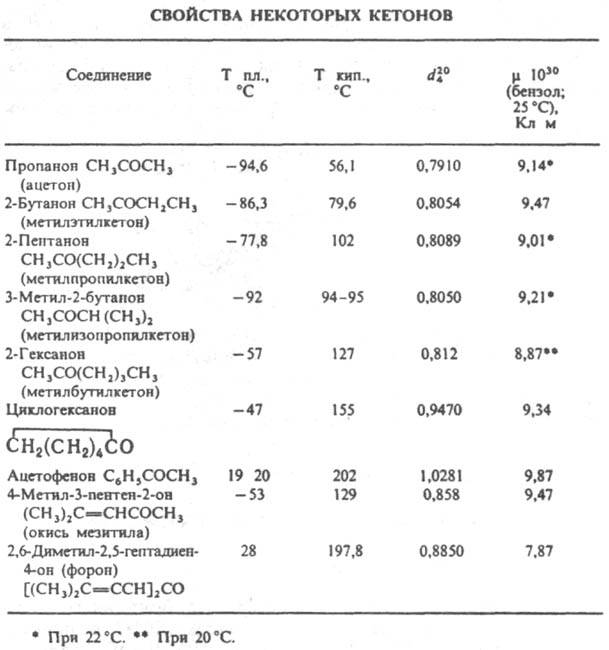
В ИК спектрах К. характеристич. полосы поглощения валентных колебаний группы С=О лежат в области частот (vC=O) 1720-1700 см-1 (алифатич. К.), 1820-1700см-1 (циклич. К.), причем vC=O возрастает с увеличением напряженности цикла. При сопряжении группы С=О с кратными связями или арилом vC=O снижается на 20-40 см-1. Хим. сдвиги протонов a-метиленовых групп К. в спектрах ПМР находятся в области 2-3 м.д., а метиленовой группы, соседней с двумя карбонилами (для дикетонов), - в области 3-4 м. д.; хим. сдвиги группы 13С=О в спектрах ЯМР 13С лежат в области 200-220 м.д. Электронные спектры К. содержат типичные полосы с lмакс 270-300 нм (e 15-20), отвечающие п:p-переходу. Для сопряженных моно-, ди- и полиеновых К. наиб. характерны полосы поглощения в области p:p* - переходов, lмакс 200-300 нм (e 10000 и выше). В масс-спектрах К. имеются пики, соответствующие a-разрыву молекулы К., причем предпочтительно отщепляется большая алкильная группа. Так, масс-спектры алифатич. метилкетонов содержат наиб. интенсивный пик с т/z 43 (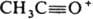 ). Для метилкетонов характерен также b-разрыв мол. иона с миграцией Н от g-атома С (перегруппировочный пик с m/z 58).
По степени окисленности К., как и альдегиды, занимают промежут. положение между спиртами и к-тами, что во многом определяет их хим. св-ва. К. восстанавливаются до вторичных спиртов гидридами металлов, напр. LiAlH4 или NaBH4, водородом (кат. - Ni, Pd), изопропаволом в присут
алкоголята Аl (р-ция Меервейна - Понндорфа - Верлея). При восстановлении К. натрием или электрохимически (катодное восстановление) образуются пинаконы:
). Для метилкетонов характерен также b-разрыв мол. иона с миграцией Н от g-атома С (перегруппировочный пик с m/z 58).
По степени окисленности К., как и альдегиды, занимают промежут. положение между спиртами и к-тами, что во многом определяет их хим. св-ва. К. восстанавливаются до вторичных спиртов гидридами металлов, напр. LiAlH4 или NaBH4, водородом (кат. - Ni, Pd), изопропаволом в присут
алкоголята Аl (р-ция Меервейна - Понндорфа - Верлея). При восстановлении К. натрием или электрохимически (катодное восстановление) образуются пинаконы:
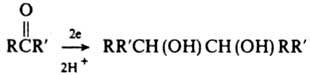
При взаимод. К. с амальгамированным Zn и конц. НСl (р-ция Клемменсена) или с гидразином в щелочной среде (р-ция Кижнера - Вольфа) группа С=О восстанавливается до СН2.
В отличие от альдегидов, многие К. устойчивы при хранении к действию О2. К., содержащие a-метиленовую группу, окисляются SeO2 до 1,2-дикетонов более энергичными окислителями, напр. КМnО4 - до смеси карбоновых к-т (см. Попова правило). Циклич. К. при взаимод. с HNO3 или КМnО4 подвергаются окислит. расщеплению цикла, напр. из циклогексанона образуется адипиновая к-та. Линейные К. окисляются надкислотами до сложных эфиров, циклические - до лактонов (р-ция Байера - Виллигера).
К., содержащие a-атомы Н, относятся к СН-кислотам средней силы (рКа 10-20). Для них характерно превращ. в енолы или енолят-анионы:
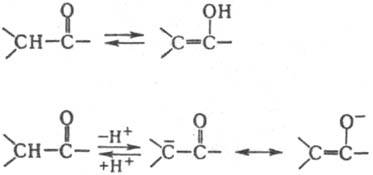
На этом основана способность таких К. реагировать как С-или О-нуклеофилы. Концентрация енольной формы зависит от строения К. и составляет (в %): 0,0025 (ацетон), 2 (циклогексанон), 80 (ацетилацетон). Енолизация катализируется к-тами и основаниями. К. образуют продукты замещения a-атомов Н при галогенировании действием Вr2, N-бромсукцинимидом, SO2Cl2, при тиилировании дисульфидами. При алкилировании и ацилировании енолятов К. образуются либо продукты замещения a-атомов Н в К., либо О-производные енолов. Большое значение в орг. синтезе имеют альдольная и кретоновая конденсации, напр.:
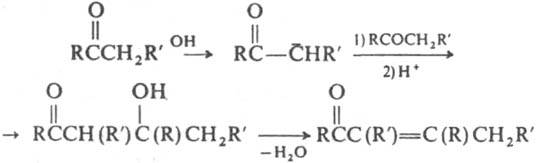
При конденсации с альдегидами К. реагируют гл. обр. как СН-кислоты, напр. из К. и СН2О в присут. основания получают a,b-ненасыщенные К.:
RCOCH3 + СН2О : RCOCH=CH2 + Н2О
Вследствие полярности карбонильной группы
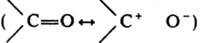 К. могут вступать в р-ции как С-электрофилы, напр. при конденсации с производными карбоновых к-т (конденсация Штоббе, р-ция Дарзана и т. п.):
К. могут вступать в р-ции как С-электрофилы, напр. при конденсации с производными карбоновых к-т (конденсация Штоббе, р-ция Дарзана и т. п.):
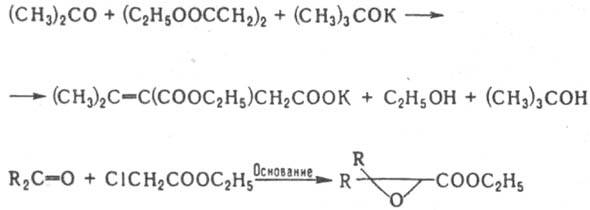
Особенно легко нуклеоф. атаке подвергаются a,b-непределъные кетоны, но в этом случае атакуется двойная связь (р-ция Михаэля), напр.:
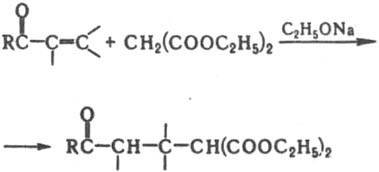
При взаимод. с илидами Р (алкилиденфосфоранами) К. обменивают атом О на алкилиденовую группу (р-ция Виттига):
R2C=O + Ph3P=CHR' : R2C=CHR' + Ph3PO
С циклопентадиеном К. образуют фулъвены, напр.:
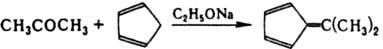
При конденсации К. с гидроксиламином получаются кетоксимы R2C=NOH, с гидразином - гидразоны R2C=N— —NH2 и азины R2C=N—N=CR2, с первичными аминами - Шиффовы основания R2C=NR', со вторичными аминами - енамины. К. способны присоединять по карбонильной группе воду, спирты, бисульфит Na, амины и др. нуклеофилы, хотя эти р-ции протекают не так легко, как в случае альдегидов.
Поскольку в спиртовых р-рах равновесие между К. и его полукеталем сильно смещено влево, получить кетали из К. и спиртов трудно:
RCOR' + R:OH D RR'C(OH)OR:
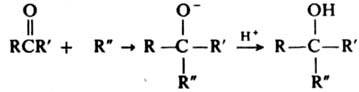
Для этой цели используют р-цию К. с эфирами ортомуравьиной к-ты. К. взаимод. с С-нуклеофилами, напр. с литий-, цинк- или магнийорг. соед., а также с ацетиленами в присут. оснований (р-ция Фаворского), образуя третичные спирты:
В присут. оснований к кетонам присоединяется HCN, давая a-гидроксинитрилы (циангидрины):
R2C=O + HCN : R2C(OH)CN
При катализе к-тами К. реагируют как С-электрофилы с ароматич. соед., напр.:
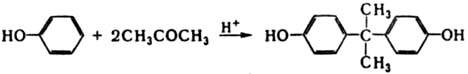
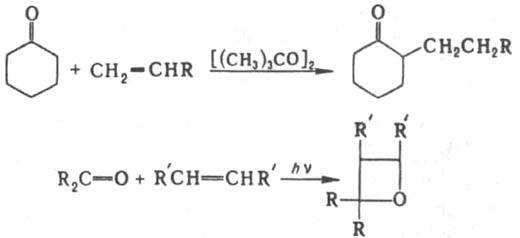
Гомолитич. присоединение К. к олефинам приводит к a-алкилзамещенным К., фотоциклoприсоединение к оксетанам, напр.:
К. играют важную роль в метаболизме в-в в живых организмах. Так, убихинон участвует в окислит. - восстановит. р-циях тканевого дыхания. К соед., содержащим кетонную группу, относятся нек-рые важные моносахариды (D-фруктоза и др.), терпены (ментон, карвон), компоненты эфирных масел (камфора, жасмон), прир. красители (индиго, ализарин, флавоны), стероидные гормоны (кортизон, прогестерон).
Общие пром. методы синтеза К.- каталитич. окисление насыщ. углеводородов и олефинов кислородом, а также дегидрирование и окислит. дегидрирование вторичных спиртов. К. синтезируют также окислит. расщеплением третичных 1,2-гликолей действием Рb(ОСОСН3)4 или НIO4, пиролизом Са- или Ва-солей карбоновых к-т, пропусканием паров к-т над оксидами Со или Тh, взаимод. зфиров или ортоэфиров карбоновых к-т и ацилгалогенидов с реактивами Гриньяра или кадмийорг. соед., ацилированием арома-тич. соед. (р-ция Фриделя-Крафтса), гидролизом геминальных дигалогенидов и др.
К. применяют как р-рители, экстрагенты, для синтеза полимеров, пестицидов, стабилизаторов, фотоматериалов, лек. и душистых в-в и др. См. также Ацетон, Ацетофенон, Бензофенон, Метилизобутилкетон, Метилэтилкетон, Циклогексанон и др.
О специфич. св-вах дикетонов см. Дикарбонильные соединения.
Лит. Бюлер К., Пирсон Д.. Органические синтезы, пер. с англ., ч. 2, М„ 1973; Общая органическая химия, пер. с англ., т. 2, М., 1982, с. 570-692, 765-847; The chemistry of the carbonyl group, v. 1, ed. by S. Patai, L. N. Y. Sydney, 1966; там же, v. 2, ed by J. Zabicky, L, 1970; Houben Weyl, Methoden der organischen Chemie, Bd 7/2a, 2b, 2c Ketone (TI 1 3), 4 Aufl, Stuttg., 1973 77.
М. Г Виноградов.