


Газовая К. х. Эффективность колонок описывается ур-нием Голея с учетом влияния сжимаемости подвижной фазы:
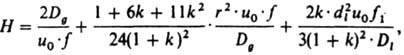
где Н - высота, эквивалентная теоретич. тарелке, Dд - коэф. диффузии хроматографируемого в-ва в газовой фазе при давлении на выходе из колонки, и0 - линейная скорость газа-носителя на выходе из колонки, u0 = L/tM.f1, где L - длина колонки, tM - время удерживания несорбирующегося в-ва, f = f1/f2, где f1 = 3/2(P2i,0 - 1)/(Р3i,0 - 1) - фактор градиента давления при усреднении по длине колонки, f2 = 4/3(P3i,0 - 1)/(P4i,0 - 1) - фактор градиента давления при усреднений по времени пребывания газа в колонке: Pi.0 = Pi/P0, где Pi - давление на входе, Р0 - давление на выходе, k - коэф. емкости колонки, r - внутр. радиус, dl - эффективная толщина пленки НЖФ, Dl - коэф. диффузии хроматографируемого в-ва в НЖФ. Первый член ур-ния отражает размывание хроматографич. зоны, обусловленное продольной диффузией в-ва в газе-носителе, второй - размывание, обусловленное параболич. изменением скоростей по сечению полой колонки и конечной скоростью массопередачи в газовой фазе, третий - размывание, обусловленное конечной скоростью массопередачи в НЖФ. Для характеристики удерживания хроматографируемых соед. с целью их идентификации в К. х. используют относит. величины: индекс Ковача I (логарифмич. индекс удерживания), линейный индекс удерживания IL и относит. удерживание r:
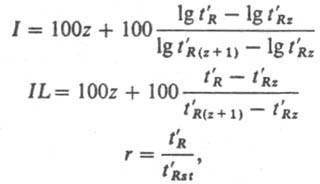
где t'R, t'Rz, t'R(z+1), t'Rst - соотв. приведенные времена удерживания хроматографируемого в-ва, нормального парафина с числом углеродных атомов z, нормального парафина с числом углеродных атомов (z + 1) и в-ва сравнения. В К. х. удерживание определяется не только сорбцией в-в в НЖФ, но и сорбцией на пов-стях раздела газ - НЖФ и НЖФ - стенка колонки (или твердый носитель). Напр., r для определения констант распределения летучих соед. в системе газ - НЖФ описывается ур-нием:
r = t'R/t'Rst = r0 + а(1/kсr),
где r0 = К/Кst - величина инвариантного относит. удерживания, К и Кst - коэф. распределения НЖФ-газ, для исследуемого соед. и для в-ва сравнения, kсr - коэф. емкости соед., используемого как стандарт для характеристики колонок с разл. содержанием НЖФ.
Низкое сопротивление потоку газа-носителя в полых колонках позволяет на стандартном хроматографич. оборудовании после модификации устройства для ввода пробы и детектора использовать классич. капиллярные колонки большой длины (50-300 м), высокой удельной (2000-4000 теоретич. тарелок/м) и общей эффективности (100-300 тыс. теоретич. тарелок).
Hаиб. широко в К. х. используют кварцевые и стеклянные колонки (в частности, для анализа полярных и неустойчивых соед.); применяют также колонки из нержавеющей стали, латуни, никеля, полимеров и др. материалов. В циркуляц. газовой хроматографии на стеклянных капиллярных колонках реализуют разделения, эквивалентные по эффективности 15 млн. теоретич. тарелок и выше.
Малое кол-во пробы в К. х. (до 10-4-10-8 г) существенно осложняет ее ввод. Различают 3 главных способа дозирования: с делением потока, без деления и прямое дозирование. В последнем способе жидкая проба вводится в колонку без предварит. испарения. Пробу в избытке легколетучего р-рителя дозируют в колонку, т-ра к-рой ниже т-ры кипения р-рителя; при этом происходит резкое увеличение емкости фазы в начале колонки и концентрированна "тяжелых" (по отношению к р-рителю) примесей.
Детектирование в-в в К. х. осуществляется с помощью высокочувствит. детекторов: пламенно-ионизационного (спец. конструкции или с применением вспомогат. газа), электронозахватного, натрийтермоионного, фотоионизационного и др. Для регистрации разделения ряда в-в, напр., неорг. газов, используют микрокатарометр спец. конструкции.
К. х. применяется для разделения сложных многокомпонентных смесей, особенно широко для разделения углеводородов, спиртов, фенолов и т.д., липидов, стероидов, пестицидов и др., летучих соед. с близкими св-вами, в т. ч. геом. и оптич. изомеров, изотопов, напр. D2, H2 и DH, 16О2 и 18О2, изотопнозамещенных молекул орг. в-в, напр. С6Н6 и C6D6. Использование полых капиллярных колонок с внутр. диаметром 0,3-1,0 мм с толстой пленкой НЖФ в комбинации с простейшей системой ввода пробы без деления потока перспективно в качеств. и количеств. анализе, в анализе легко- и среднесорбируемых соед., а также в анализе следов, рутинном анализе и в пром. хроматографии на потоке.
Капиллярные насадочные колонки обладают нек-рыми преимуществами перед полыми колонками: более высокой удельной эффективностью (10-30 тыс. теоретич. тарелок/м); простотой реализации газо-адсорбц. варианта хроматографии; возможностью эффективного разделения и экспрессного аналит. определения легко- и среднесорбируемых соед. (включая неорг. газы); возможностью использования в термостате колонок малого объема (миниатюризация газохроматографич. аппаратуры). Осн. препятствие для широкого применения таких колонок в существующих приборах для газовой хроматографии - значит. сопротивление потоку газа-носителя.
Жидкостная К. х. При работе с несжимаемой подвижной фазой зависимость приведенной высоты h, эквивалентной теоретич. тарелке, от приведенной скорости n для полых капиллярных колонок выражается ур-нием:
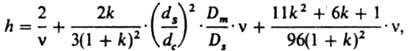
где h = H/dc, n = udc/Dm, dc - внутр. диаметр колонки, Dm - коэф. диффузии хроматографируемого в-ва в подвижной (жидкой) фазе, и - линейная скорость подвижной фазы, ds - толщина слоя неподвижной фазы, Ds - коэф. диффузии хроматографируемого в-ва в неподвижной фазе, k - коэф. емкости колонки.
Для ввода пробы используют метод остановки потока, введение пробы непосредственно в колонку и с помощью микрокрана. Для детектирования используют спектральные (в УФ и ИК областях), масс-спектральные, пламенно-ионизационные и электрохим. детекторы. Объем кюветы или ячейки детектора должен соответствовать объему колонки (в К. х. - несколько нл).
В К. х. на насадочных колонках реализованы осн. варианты жидкостной хроматографии: прямо-фазный, обращенно-фазный, ионный, эксклюзионный. Она успешно используется для определения полициклич. ароматич. углеводородов, аминов, нуклеозидов, фенолов, полимеров дейтерированных и прир. соед., лек. ср-в и т.д. Однако жидкостная К. х., в отличие от газовой, еще не нашла широкого применения.
Флюидная К. х. основана на использовании в качестве подвижной фазы СО2, N2O и др. газов, сжатых до сверхкритич. состояния (флюиды), и полых капиллярных колонок с внутр. диаметром 25-100 мкм. Растворяющая способность флюида сопоставима с растворяющей способностью подвижной фазы в жидкостной хроматографии, а значение коэф. диффузии растворенных во флюиде в-в на 2-3 порядка выше, чем в жидкостной хроматографии. Это св-во флюида в сочетании с относительно низкой его вязкостью позволяет увеличить эффективность разделения. При разделении многокомпонентных смесей в-в коэф. распределения и время элюирования регулируют программированием плотности флюида. Для детектирования применяют универсальный к орг. в-вам пламенно-ионизац. детектор, оптич. спектральный детектор или масс-спектрометр.
Сверхкритич. флюидная хроматография расширяет границы применения К. х. в области анализа смесей труднолетучих и термолабильных соед., особенно соед. прир.
происхождения (тяжелые фракции нефти, лек. ср-ва, смеси олигомеров и др.) с мол. м. до 3000.
Создатель газовой К. х. и теоретич. основ метода -М. Голей. В газовой и жидкостной хроматографии полые капиллярные колонки предложены соотв. М. Голеем в 1957 и Г. Нота, Дж. Марино, В. Буопокоре, А. Баллио в 1970.
Лит.: Руденко Б. А., Капиллярная хроматография, М., 1978; Березкин В. Г. [и др.], "Успехи химии". 1978. т. 47. в. 10. с. 1875-1903: Дженнингс В., Газовая хроматография на стеклянных капиллярных колонках, пер. с англ., М., 1980; Березкин В. Г., Золоторев П. П., "Успехи химии", 1984, т. 53. в. II, с. 1891-1924; Тесаржик К., Комарек К., Капиллярные колонки в газовой хроматографии, пер. с чеш., М., 1987; Microcolumn separations, ed. by M. Novotny and Daido Ishi. Amst., 1985. В. Г. Березкин.