


ВИНИЛХЛОРИД (хлористый винил, хлорэтен, монохлорэтилен) СН2—СНС1, мол. м. 62,50; бесцв. газ с эфирным запахом. Длина связей: С—С1 0,169 нм, С—С 0,138 нм; энергия связей (кДж/моль): С—С1 338,9, С—С 599,98.
Свойства. Т. пл. -158,4°С, т. кип. -13,8°С; d4-20
0,983,
d420 0,911 (жидкость), плотность по воздуху
2,17, nD10 1,4046, nD20
1,3700; давление пара (кПа): 0,13 (-105,6°С), 13,33 (-53,2°С), 172 (0°С),
337 (20°С), 2518 (100°С); tкрит 158,4 °С, pкрит5,34
МПа, dкрит 0,370 г/см3;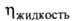 (мПа*с) 0,334 (-40°С), 0,256 (-1(10°С), 0,180 (20°С),
(мПа*с) 0,334 (-40°С), 0,256 (-1(10°С), 0,180 (20°С),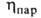 (мкПа*с) 9,20 (-20°С), 10,71 (20°С), 13,71 (100°С);
(мкПа*с) 9,20 (-20°С), 10,71 (20°С), 13,71 (100°С);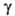 (мН/м) 22,3 (-20°С), 16,9 (20°С); Сp, [кДж/(кг*К)] жидкого
1,146 (-20°С), 1,351 (20°С), пара 0,858 (25°С), 1,000 (100°С); теплопроводн.
жидкости при 20°С 0,138 Вт/(м*К);
(мН/м) 22,3 (-20°С), 16,9 (20°С); Сp, [кДж/(кг*К)] жидкого
1,146 (-20°С), 1,351 (20°С), пара 0,858 (25°С), 1,000 (100°С); теплопроводн.
жидкости при 20°С 0,138 Вт/(м*К);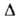 Н°исп(кДж/кг)
332,7 (-13,8°С), 298,2 (25°С), 217,6 (100°С),
Н°исп(кДж/кг)
332,7 (-13,8°С), 298,2 (25°С), 217,6 (100°С),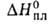 75,9 кДж/кг,
75,9 кДж/кг,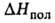 92,11 кДж/моль,
92,11 кДж/моль,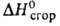 - 1198,1 кДж/моль,
- 1198,1 кДж/моль,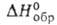 - 37,26 кДж/моль; Sо298 263,98 Дж/(моль*К)
при 25°С;
- 37,26 кДж/моль; Sо298 263,98 Дж/(моль*К)
при 25°С;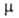 4,84*10-30 Кл*м и 3,00*10-30 Кл*м для жидкости и
пара соотв.;
4,84*10-30 Кл*м и 3,00*10-30 Кл*м для жидкости и
пара соотв.;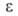 6,26 (25°С). В. хорошо раств. в обычных орг. р-рителях; р-римость в воде
0,25% (0°С), 0,11% (25°С), р-римость воды в В. 0,042 (0°С), 0,097% (20
°С).
6,26 (25°С). В. хорошо раств. в обычных орг. р-рителях; р-римость в воде
0,25% (0°С), 0,11% (25°С), р-римость воды в В. 0,042 (0°С), 0,097% (20
°С).
По двойной связи В. вступает в р-ции, типичные для олефинов; НС1 присоединяется к В. по правилу Марковникова, причем легче, чем к этилену, но с меньшей скоростью, чем к винилиденхлориду; р-ция может протекать в жидкой фазе в присут. А1С13 или FeCl3. В последнем случае возрастает селективность, но требуются повышенные т-ра (50-70°С) и давление. Продукт взаимод. НС1О с В. - хлорацетальдегид. Большое практич. значение имеет полимеризация В. и сополимеризация его с др. мономерами. Ингибиторы полимеризации - фенол или гидрохинон. В. значительно менее активен при замещении атома хлора, чем этилхлорид. Однако хлор м. б. замещен, напр., при нагр. В. в спиртовом р-ре алкоголята Na под давлением, а также при взаимод. В. с солями карбоновых к-т с образованием винилалкиловых эфиров, при конденсации с ароматич. или жирноароматич. соединениями Гриньяра в присут. галогенидов металлов типа СоС12 или СгС13. Окисляется В. до хлорацетальдегида или формальдегида, НС1 и СО, алкилируется, дегидрохлорируется в газовой фазе до ацетилена.
Получение. 1) наиб. старый метод получения В. - гидрохлорирование ацетилена
в паровой фазе - проводят при 150-220°С (кат. - активиров. уголь, пропитанный
10-15%-ным р-ром сулемы). Тщательно очищенные осушенные ацетилен и НС1
после смешения поступают в трубчатые реакторы. Тепло р-ции (145 кДж/моль)
отводится водой, циркулирующей в межтрубном пространстве. Реакц. газы промывают
водой, нейтрализуют, сушат, компримируют и подают на ректификацию, где
выделяют чистый В. 2) При получении В. из ацетилена и этилена последний
хлорируют до дихлорэтана (ДХЭ), к-рый подвергают парофазному дегидрохлорированию
в трубчатой печи при 400-550 °С до В. и НС1. Реакц. газы быстро охлаждают
("закалка"), отделяют от продуктов осмоления и разделяют: НС1 направляют
на гидрохлорирование ацетилена (по схеме метода 1), из остатка ректификацией
выделяют чистый В., непрореагировавший ДХЭ (30-50% от исходного) возвращают
на дегидрохлорирование. 3) Из прямогонного бензина В. получают по схеме:
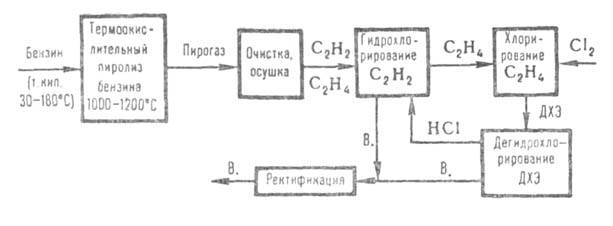
Пирогаз, содержащий по 8-10% ацетилена и этилена, очищают от смолы и
высших гомологов ацетилена и этилена, осушают и подвергают гидрохлорированию
(по схеме метода 1, только под давл. до 0,61 МПа). После выделения В. этилен
поступает на хлорирование до ДХЭ (0,51 МПа; кипящая реакц. среда), к-рый
выделяют из реакц. газов конденсацией и после ректификации дегидрохлорируют
(по схеме метода 2, только под давл. 1,0 МПа). 4) наиб. распространение
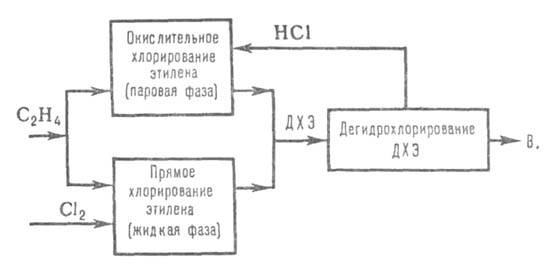
получил процесс получения В. из этилена по сбалансированной по хлору схеме
(см. ниже). Этилен примерно в равных кол-вах подают в реакторы прямого
и окислит. хлорирования. Катализатор окислит. хлорирования - СиС12
на носителе. Образовавшийся на обеих стадиях ДХЭ после очистки и сушки
объединяется, подвергается ректификации и дегидрохлорированию по схеме
метода 2 (условия дегидрохлорирования, как в методе 3). Побочные продукты
(до 100 кг на 1 т В.) в основном м. б. переработаны в перхлоруглеводороды.
Достоинства первого метода: простота технол. схемы и используемого оборудования, высокие степень превращ. исходных продуктов (99%) и выход В. (98-99%); недостатки: малая производительность одного реактора (макс. 8-10 тыс. т/год) и высокая стоимость ацетилена, из-за чего этот метод не получил широкого распространения. Самыми высокими технико-экономич. показателями обладает последний процесс благодаря большой производительности реакторов (120-250 тыс. т/год), высокой автоматизации, низкой стоимости этилена.
В лаборатории В. получают дегидрохлорированием ДХЭ спиртовым р-ром NaOH или КОН в метаноле или этаноле при 60-70 °С. Образующийся в виде паров В. сушат гранулированным NaOH и собирают при т-ре от — 25 до -30°С.
Допустимое содержание примесей в В. высшего сорта (%): ацетилена - 0,0001; ацетальдегида - 0,001; дихлорэтанов - 0,001; 1,3 - бутадиена-0,001; хлоропрена - 0,0001; прочих орг. примесей - 0,026; НС1 - 0,0001; железа - 0,0001; влаги - 0,02.
В. используют гл. обр. для произ-ва поливинилхлорида, а также разл. сополимеров (см. Винилхлорида сополимеры). М.б. сырьем для произ-ва винилиденхлорида и метилхлороформа.
Для В. т. всп. -77,8°С (в открытом приборе), -61,1°С (в закрытом), т. самовоспл. 472°С; КПВ в воздухе 3,6-33,0%, в кислороде - 4,0-70,0%. ПДК 0,1 мг/м3 (рекомендуемая).
Мировое произ-во 15 млн. т/год (1981).
Лит.: Трегер Ю. А., Пименов И. Ф., Гольфанд Е. А., Справочник
по физико-химическим свойствам хлоралифатических соединений Ci-Cj, Л.,
1973; Промышленные хлорорганические продукты. Справочник, под ред. Л. А.
Ошина, М., 1978, с. 53-70. Ю.А. Трегер.