


АДИАБАТИЧЕСКОЕ ПРИБЛИЖЕНИЕ в квантовой химии, метод анализа молекулярных систем, заключающийся в том, что в системе выделяют и раздельно описывают две или неск. подсистем, для к-рых характерные времена изменения состояния сильно различаются. Предполагается, что медленное изменение состояния одной из подсистем не влияет на состояние другой, для к-рой характерное время его изменения существенно меньше. наиб. важные применения А. п. связаны с разделением в ур-нии Шрёдингера для молекулы или кристалла переменных r и R, описывающих состояния соотв. электронов и ядер. Разница в характерных временах изменения состояния электронной и ядерной подсистем при их взаимод. обусловлена существенным (в тысячи раз) различием масс ядер и электронов.
В рамках А. п. считается, что электроны молекулы, рассматриваемые в поле фиксированных в своих мгновенных положениях ядер, могут находиться в неск. состояниях, каждому из к-рых отвечает энергия Еп (R), где и - номер состояния. Ядра же находятся в создаваемом электронами поле, усредненном по всем положениям электронов и имеющем потенциал En(R). Ф-ции En(R)представляют собой многомерные поверхности потенциальной энергии (в случае двухатомных молекул-потенциальные кривые), что позволяет связать их минимумы с равновесными геом. конфигурациями ядер и описывать молекулярную структуру с помощью таких понятий, как длины связей, валентные углы и т.п.; седловые точки на пов-сти потенциальной энергии соответствуют переходным состояниям (см. Активированного комплекса теория).
В n-м состоянии система электронов описывается электронной волновой
ф-цией Фn (r, R), к-рая зависит от координат R как
от параметров. Эта ф-ция и энергия En(R)определяются
решением т. наз. электронного ур-ния Шрёдингера
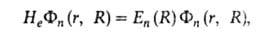
где Нe - электронный гамильтониан. Существует неск. типов А. п., различающихся выбором формы Не, но во всех случаях Не отличается от гамильтониана молекулы как целого учетом действия на ф-цию Фn (r, R)оператора кинетич. энергии ядер Т и способом разделения переменных, описывающих движение центра масс молекулы и вращение ее как целого. В простейшем случае действием Т на Фn (r, R)пренебрегают (приближение Борна-Оппенгеймера). Массы ядер в этом случае не входят в выражение для He, что позволяет объяснить в рамках А. п. хим. эквивалентность изотопов и изотопные эффекты, поскольку изотопы различаются только массами ядер.
При фиксированном электронном состоянии подсистема ядер описывается
ядерной волновой ф-цией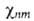 (R),
где
n и m-номера соотв. электронного и ядерного состояний. Ядерное ур-ние Шрёдингера
определяет
(R),
где
n и m-номера соотв. электронного и ядерного состояний. Ядерное ур-ние Шрёдингера
определяет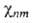 (R)и
энергетич. уровни молекулы Еnт :
(R)и
энергетич. уровни молекулы Еnт :
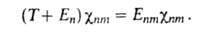
Волновая ф-ция молекулы как целого в рамках А. п. есть произведение
Фn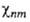 .
Энергетич. уровни Епт с разными значениями т образуют
набор колебательно-вращат. состояний молекулы, отвечающих данному электронному
состоянию.
.
Энергетич. уровни Епт с разными значениями т образуют
набор колебательно-вращат. состояний молекулы, отвечающих данному электронному
состоянию.
Для перехода от волновой ф-ции, найденной в А. п., к точным волновым ф-циям молекулы необходимо учитывать согласованность электронных, колебат. и вращат. движений молекулы с помощью т. наз. неадиабатических поправок. Надежность А. п. и соответственно величины этих поправок определяются соотношением характерных времен te и tN изменения состояний электронной и ядерной подсистем.
Применение А.п. правомерно, если так называемый параметр Месси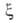 =
tN/te
=
tN/te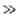 1.
Значение te оценивают обычно по энергии
1.
Значение te оценивают обычно по энергии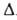 Ееквантовых
переходов с изменением электронного состояния молекулы: te
=
Ееквантовых
переходов с изменением электронного состояния молекулы: te
=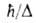 Еe,
где
Еe,
где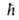 -постоянная
Планка. Применение аналогичного соотношения для оценки tn показывает,
что при равновесных конфигурациях ядер А. п., как правило, хорошо выполняется,
т.к. энергии электронных переходов в 10-10000 раз больше энергий переходов
с изменением лишь колебательно-вращат. состояния. В этом случае неадиабатич.
поправки малы по сравнению с характерными для хим. процессов энергиями.
При рассмотрении реакц. способности tN часто оценивают
как отношение расстояния между частицами, на к-ром заметно меняется электронная
энергия, к скорости движения ядра (или группы ядер). Поэтому при изучении
процессов, характеризуемых большими скоростями движения ядер, неадиабатич.
эффекты существенны. Их необходимо учитывать при анализе вырожденных или
почти вырожденных электронных состояний, когда становятся важны электронно-колебат.
взаимодействия.
-постоянная
Планка. Применение аналогичного соотношения для оценки tn показывает,
что при равновесных конфигурациях ядер А. п., как правило, хорошо выполняется,
т.к. энергии электронных переходов в 10-10000 раз больше энергий переходов
с изменением лишь колебательно-вращат. состояния. В этом случае неадиабатич.
поправки малы по сравнению с характерными для хим. процессов энергиями.
При рассмотрении реакц. способности tN часто оценивают
как отношение расстояния между частицами, на к-ром заметно меняется электронная
энергия, к скорости движения ядра (или группы ядер). Поэтому при изучении
процессов, характеризуемых большими скоростями движения ядер, неадиабатич.
эффекты существенны. Их необходимо учитывать при анализе вырожденных или
почти вырожденных электронных состояний, когда становятся важны электронно-колебат.
взаимодействия.
А. п. лежит в основе практически всех представлений совр. теоретич. химии о строении молекул, хим. связи, реакц. способности, динамике элементарного акта хим. р-ции, природе фотохим. процессов. В рамках А. п. сформированы осн. понятия и методы интерпретации эксперим. данных в молекулярной спектроскопии, электронографии, рентгенографии и др. областях структурной химии.
Лит.: Давыдов А.С, Квантовая механика, М.. 1963: Кондратьев В.Н.,
Никитин Е.Е., Кинетика и механизм газофазных реакций, М., 1975; Браун П.А.,
Киселев А.А., Введение в теорию молекулярных спектров, Л.. 1983. В.
И. Пупышев.